Abstract
Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome is a rare, severe autosomal dominant genetic disorder caused by heterozygous variants in the SON gene. This syndrome characterized by intellectual disability, brain malformation, facial dysmorphism, musculoskeletal abnormalities, and some visceral malformations. There are only 33 cases reported to date since the first report in 2015.
Herein, we present the first Tunisian patient with ZTTK syndrome due to a mutation in SON gene. The clinical features of our patient were strikingly similar to previously reported cases.
Introduction
Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome is a severe multi system developmental disorder, first recognized in 2015 by Zhu et al. [1]. ZTTK syndrome is an autosomal dominant hereditary disease caused by de novo heterozygous loss-of-function mutations in SON gene [2]. SON is located on the human chromosome region 21q22.11 [2,3]. The mutations found to be associated with ZTTK syndrome are mostly frame shift mutations and nonsense substitutions generating a premature termination cod on and transcripts of the mutant gene seem to be degraded due to Nonsense-Mediated mRNA Decay (NMD) [3]. SON haploinsufficiency results in reduced mRNA expression and abnormal RNA splicing of many genes, which are necessary for neural cell migration, metabolic processes, renal and brain development [4,5]. It was further characterized as a congenital anomaly syndrome of delayed psychomotor development, intellectual disability, brain malformation, epilepsy, characteristic facial features, musculoskeletal abnormalities, mucocutaneous manifestations and less common visceral malformations [1–6]. Till now, 60 individuals with pathogenic variants in SON were reported [7].
Here in, we report the first Tunisian ZTTK syndrome. The clinical manifestations of our patient were similar to those in previously reported cases.
Case Report
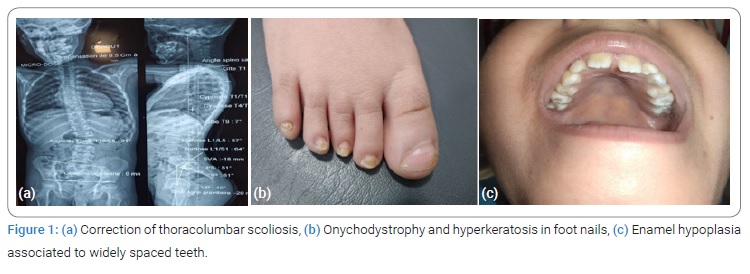
A 9-year-old boy is followed for moderate intellectual disability, developmental delay and multiple congenital abnormalities. Parents were healthy and unrelated. Pregnancy course was uneventful. Delivery was at term. He was eutrophic at birth g, without a history of asphyxia. His psychomotor and language development were significantly delayed at the age of 2. He had a medical story of thoracolumbar scoliosis for which he had scoliosis braces (Figure 1a). He presented a first seizure at the age of 3 years when he was started on valporic acid. EEG analysis showed changes in the left mid-temporal area, activated by sleep. Later on, he presented Attention Deficit Hyperactivity Disorder (ADHD) with poor school performances associated to refractive error.
On examination, body weight was 30 kg (50e percentile) and height was 126 cm (25 percentile). He showed distinctive dysmorphic feature: facial asymmetry, micrognathia, depressed nasal bridge, short philtrum, ogival palate, and enamel hypoplasia associated to widely spaced teeth (Figure 1b and 1c). Onychodystrophy and hyperkeratosis were noticed in all of the ten feet nails (Figure 1b). He presented proximal muscle weakness with generalized joint laxity.
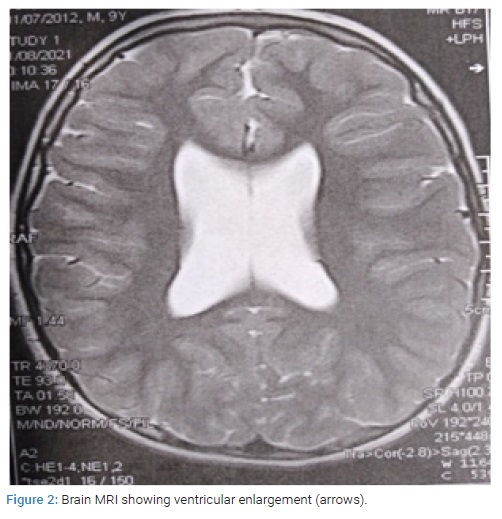
Psychologic examination revealed intellectual disability, inattention and hyper activity with moderate intellectuel disability. At the age of 9, the Intelligence Quotient (IQ) was about 55. The brain MRI showed parallel bilateral lateral ventricles, ventricular enlargement, and thin corpus callosum (Figure 2). The cardiac ultra sound concluded to moderate pulmonary artery stenos is without hemodynamic effects.
Before Whole-Exome Sequencing (WES), the following genetic tests were performed yielding normal results: karyotype, as well as array CGH. Array-CGH as say was performed and any structural chromosomal aberration was revealed. WES was performed on the prob and. We identified a heterozygous de novo frame shift variant in the SON gene, NM_138927.2: c. 5753_5756del p. (Val1918Glufs*87) which creates a shift in the reading frame starting at codon 1918. The new reading frame ends in a stop codon 86 positions downstream. The result is consistent with the genetic diagnosis of autosomal dominant ZTTK syndrome.
Discussion
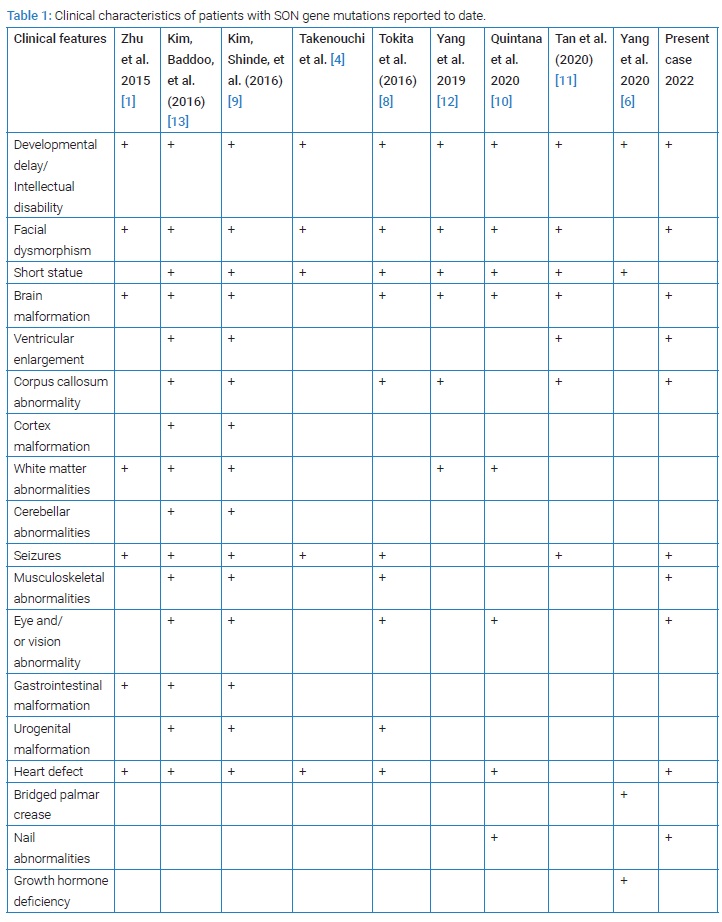
We present a rare case of ZTTK syndrome with symptoms similar to those described by other authors as shown in (Table 1) and in whom the pathogenic variants in the SON gene were identified. Variant analysis identified a heterozygous pathogenic variant c.5753_5756del p.(Val1918Glufs*87) in the SON gene. This variant has previously been described as disease causing for developmental delay, seizure disorders, macrocephaly and brain white matter abnormalities by Zhu et al. [1], Tokita et al. [8], Kim et al. [9].
Facial Dysmorphism
Case report displayed dysmorphic features: Facial asymmetry, micrognathia, ogival palate, depressed nasl bridge, short philtrum and enamel hypoplasia associated to widely spaced teeth. These abnormalities have been reported in several studies [1,4,10]. Distinctive facial features include also mid face retraction, low-set ears, down slanting palpebral fissures, deep-set eyes, horizontal eye-brows [10]. Enamel hypoplasia was first described by Takenouchi et al. in two patients with the disorder [4].
Onychodystrophy and hyperkeratosis in feet nails: Case report showed Onychodystrophy and hyperkeratosis in all of the ten feet nails. In 2020, Lucía Quintana Castanedo et al. were the first who described nails abnormalities which can provide a useful diagnostic clue [10].
Neurological Abnormalities: Similar to the all previous reported cases of ZTTK syndrome, intellectual disability and developmental delay were found in our children. Poor school performances and Attention Deficit Hyperactivity Disorder (ADHD) are also noticed in this index case.
Brain malformations (ventricular enlargement, thin corpus callosum, abnormal cerebral cortical gyration, arachnoid cyst, cerebellar dysplasia, and/or white matter abnormality) were observed in 85.9% of the subjects [6,11,12]. Neurological features such as hyper-or hypotonia, epilepsy or other EEG abnormalities, and autism spectrum disorder occurred in 62.0% of the subjects [6].
The index case underwent brain MRI which showed parallel bilateral lateral ventricles, ventricular enlargement, and thin corpus callosum. EEG analysis showed changes in the left mid-temporal area, activated by sleep. Our patient had his first seizure at around age 3.
Cardiovascular System
One-third of patients were diagnosed with a variety of heart defects among which patent Ductus Arteriosus (PDA), Atrial Septum Defect (ASD) or Ventricular Septum defect (VSD) defects were most frequently observed [2,5,7,11]. Fortunately, the cardiac ultra sound of the index case showed a moderate pulmonary artery stenosis without hemodynamic effects.
Musculoskeletal Anomalies
Musculoskeletal abnormalities (joint laxity, scoliosis, or kyphosis, hemi vertebrae, cubitus valgus, contractures, small hands and feet, and/or abnormal ribs) were identified in 77.3% of the subjects [1,5,6]. Evenly, proximal muscle weakness, thoracolumbar scoliosis, generalized joint laxity were noticed in the index case. Short stature occurred in 51.8% of the cases [6]. As for our patient, body high was 25e percentile. Growth hormone deficiency was once reported by Yang et al. [6].
Eye disorders: Eye and/or vision abnormalities, have also been reported in approximately 69.6% of the cases [6]. Strabismus was the most frequently described change [5]. Others include hyperopia, cortical visual impairment, and optic atrophy [5,11]. Mean while, our patient had refractive error.
Other clinical manifestations: Other manifestations include urogenital malformations (single kidney, horseshoe kidney, and kidney dysplasia), intestinal atresia and feeding difficulties [1,5,6,13]. Immunoglobulin deficiency and abnormal coagulation also occur in several patients which leads to increased frequency of infections, especially of middle ear, urinary, and respiratory tracts (lower and upper respiratory tract) [5,6].
Conclusion
The present study reports a child with ZTTK syndrome carrying the c.5753_5756del mutation of SON (NM_138927.2). A ZTTK syndrome case caused by a SON mutation was reported for the first time in the Chinese population. Also, we emphasize the importance of WES for early diagnosis of ZTTK syndrome, which can speed up the diagnostic procedure, sparing patients from unnecessary investigations.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
Keywords
Zhu-Tokita-Takenouchi-Kim syndrome; SON gene; Multiple-system disorder
Cite this article
Haddad S, Amdouni R, Mezghani F, Jebebli E, Rhayem S, Fdhila F, et al. Zhu-Tokita-Takenouchi-Kim syndrome: clinical and genetic analysis of the first Tunisian case caused by De Novo mutation C. 5753_5756 Del in the SON Gene. Glob J Pedia. 2022;1(1):1–4.
Copyright
© 2022 Haddad Samir This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
