Predicting Biopterin Responsive Cases by Studying Phenylalanine Hydroxylase Mutations in Children Diagnosed with Phenylketonuria in Syria
* Diana Alasmar;
-
* Diana Alasmar: Endocrine and Metabolic Diseases Unit, Children Hospital, Damascus University, Damascus, Syria.
-
Jun 29, 2022 |
-
Volume: 1 |
-
Issue: 2 |
-
Views: 1558 |
-
Downloads: 2151 |
Abstract
Aim: Phenylketonuria occurs in patients with phenylalanine hydroxylase deficiency, and some mutations are responsive to Biopterin (BH4) therapy, which reduces the need for the use of hydrolysates and diet foods. This study describes the clinical and genetic features of 35 independent, authentic Syrian cases, in order to identify the dominant genotypes and predict response to BH4.
Methodology: A prospective study of a population of patients diagnosed with phenylketonuria and those attending the Metabolic Clinic at Children's University Hospital in Damascus. The study of the phenylalanine hydroxylase gene was conducted in the Atomic Energy Authority in the Syrian Arab Republic, and the clinical classification and predicting Phenotype was carried out according to the Guldberg classification and BioPKU database.
Results: The study included 35 cases of phenylalanine hydroxylase deficiency, 65% had a classic pattern. We Identified 16 different mutations, 75.6% missense, 33.4% splice, 9% frameshift. The most frequent mutations were p.R261Q (12.6%) then p.R243Q, p.F55>Lfs, and IVS2+5G>C in 8.75% each. Some rare mutations were found in pS310F, Y387H, IVS9+G>A, and p.D151G, the latter of which the database did not provide information on.
The genotype-phenotype correlation was positive in 80.3%, response to biopterin was probable in 22.9% of cases.
Conclusion: The diagnosis of the genotype is an important step that allows for directing treatment and predicting future prognosis for PKU patients.
Introduction
Phenylalanine Hydroxylase (PAH) masters the metabolism of Phenylalanine (Phe) to tyrosine in the presence of the molecular oxygen and cofactor tetrahydrobiopterin (BH4) [1,2]. PHA deficiency causes Phenylketonuria (PKU). PKU is an autosomal recessive disease that manifests with neurological symptoms with varying degrees, including psychomotor delay, mental delay, convulsions, decreased growth and microcephaly, sometimes accompanied by digestive symptoms such as nausea and vomiting, and skin symptoms such as eczema and pale skin [3,4]. PKU Treatment depends on the restriction of dietary resources of Phe with or without Biopterin (BH4) administration that has varying degrees of response [5].
In general, individuals who maintain levels of Phe in the range of (300 to 600) μmol/L ((5 to 10) mg/ dl) on a normal diet are classified as having Mild Hyperphenylalaninemia (MHP). These individuals generally do not need diet therapy [5].
The gene encoding human PAH is located on chromosome 12q23.2 and contains 13 exons and 12 introns of 90 kb size [6].
More than 1000 mutations have been published in the PAH database www.pahdb.mcgill.ca with most families carrying compound heterozygote mutations for two different mutant alleles [7].
The diversity of phenotypes shows that significant heterogeneity in the mutant alleles is behind the occurrence of phenylketonuria and the associated forms of hyperphenylalaninemia [1]. The genotype phenotype correlation has been announced in some European peoples they are of Middle Eastern origin, but there are few reports from the Middle East, including Egypt, Israel, Morocco, Tunisia [8–11].
The structure and activity of the PAH varies according to the mutation type (missense, nonsense, splice site, small or large insertions and deletions) and the position (regulatory, catalytic, or tetramerization domain). The activity of mutant protein ranges from 0% to almost 100% compared to normal PAH enzyme Waters et al. 1998 [12].
There is 4 enzymes recycle BH4, the cofactor of PAH, which is also the cofactor for both tyrosine hydroxylase and tryptophan hydroxylase, the defect of BH4 cycle leads to the deficiency of serotonin and dopamine in addition to hyperphenylalanine and neurological deterioration occurs regardless of phenylalanine levels and application Diet Therefore, conditions associated with biopterin metabolism are described as Malignant PKU or Malignant HPA (mgHPA) [7,13].
Tetrahydrobiopterin (BH4) trial is becoming a valid option for the dietary treatment of PKU It relies on the observation that pharmacological doses of BH4 can reduce Phe levels Kure et al. 1999 [14], “BH4- sensitive PKU”.
Many studies analyzed the correlation of genotype and patient’s responsiveness to BH4 and made an attempt to predict the BH4-responsiveness on the basis of genotype as Zurfluh et al. 2008 [15]; Karacic et al. 2009 [16]; Rivera et al. 2011[17]; Sterl et al. 2012 [18].
Actually, BH4 loading test is the definitive diagnostic test for cofactor sensibility. BH4-responsiveness is defined as the response to the oral administration of BH4 ((10 to 20) mg/kg body weight) by lowering their blood Phe levels by at least 30% within 8 hr to 24 hr [13]. However, estimation based on genotyping is important to know if the frequency of BH4 responsive mutations in a population is high enough to consider BH4 in their treatment policy.
This study describes the clinical and genetic features of 35 independent, authentic Syrian cases, in order to update genotype-phenotype correlation and predict the response to BH4.
Patients and Methods
This study included all PKU patients followed in the Metabolic Clinic at Children's University Hospital in Damascus. Out of 101 families that were contacted only 41 patients from unrelated families of Syrian nationality had responded. Sample collection lasted for 28 months until the end of 2012. Information was collected (phe) level before starting the diet. In the event that there is more than one case in the family, the analysis was performed for one individual only.
PAH gene was studied in the laboratory of the Atomic Energy Authority in the Syrian Arab Republic, for the six most common mutations p.E280K, IVS10-11GNA, p.P281L, IVS11+1GNC, p.R26Q, p.S310F and In case of negativity, a study was conducted to study the PAH gene sequence using RFLP and Gene Sequencing technology.
The diagnosis of PHA deficiency was based on the presence of a confirmed mutation in the PHA gene, and the negative cases were excluded as these cases must be studied for a defect in the metabolism of BH4 cofactor [7].
Clinical classification of patients according to the level of phenylalanine elevation before treatment and estimation of the nutritional tolerance of phenylalanine from the food intake was performed into four groups according to Guldberg et al. [19], classical phenylketonuria (cPKU), Phe level > 1200 µmol/l with Phe tolerance Diet (250 to 350) mg/day, metabolic activity < 1%, moderate (mPKU): Phe level 600 μmol/L–1200 μmol/L with a dietary Phe tolerance of (350 to 400) mg/dL/day and metabolic activity 1% to 5%, mild (miPKU) Phe level between (369 to 600) µmol/l with afferent Phe tolerance between (400 to 600) mg/day, enzymatic activity > 5% and Mild Hyperphenylalanineemia (MHP) with Phe less than 360 µmol/l with normal dietary efficacy.
The Guldberg et al. [19] system was adopted in the clinical phenotype classification so that when two mutations were identified, each of them was given an Arbitrary Value (AV): AV = 1 for the classic cPKU mutation, AV = 2 for the mild mPKU mutation, AV = 4 for the mild miPKU, and AV = 8 for the hyperphenylalanine mutation MHP.
In the case of a combined heterozygous resulting from the meeting of two different mutant alleles, AV is the result of the addition of two mutations. The less severe mutation determines the patient's morphology, and two mutations of the same severity will cause a milder morphology than if one of them were alone in the homozygous form. The nonsense, frame shit and splice-site mutations were considered as Null mutations [19]. The Allelic Phenotype Values (APV) was calculated based Biopku db (http://www.biopku.org) assigned APVs [0 = classic PKU; 5 = mild PKU; 10 = mild hyperphenylalaninaemia]. The Genotypic Phenotype Values (GPVs) were set equal to the higher-APV allele, which was assumed to be dominant over the lower-APV allele and to determine the metabolic phenotype. GPVs for 8872 patients resulted in cut-off ranges of 0.0 to 2.7 for classic PKU, 2.8 to 6.6 for mild PKU and 6.7 to 10.0 for mild hyperphenylalaninaemia. (Sven F.AVP article) Biopku db has recorded 1282 mutations, the site were finally updated on June 06, 2021. The expected phenotype in each mutation was compared to the most important studies: Bercovich et al. [20], Danielle et al. [21], Mallolas et al. [22] and Santos et al. [23].
Estimated BH4 responsiveness
Estimated BH4 responsiveness of each mutation as a categorical variable (NR: Non-Responsive; R: Responsive; UNC: Uncertain; UNK: Unknown) was based on the work of Zurfluh et al. 2008 [18], and as indicated when analyzing the BIOPKU database (http://www.biopku.org). A mutation was considered by Zurfluh et al. (2008) [18] as associated with BH4‐responsiveness if it was present either in homozygosis or in compound heterozygosis with a known null mutation in patients that were classified as BH4‐responsive-response to the oral administration of BH4. In the BioPKU db, the indication of BH4‐responsiveness of a mutation was taken into account when the majority of listed patients carrying it either in homozygosis or in compound heterozygosis with a known null mutation was classified as BH4‐responsive [24,25].
Results
Out of the 82 studied alleles 75 mutant alleles were identified, we obtained the genotype for only 35 patients (19 F, 16 M) the other 6 cases was negative for PHA gene mutation and must be studied in order to exclude one of the deficiencies of the metabolism of BH4 coenzyme, which is not available in the country.
The mean Phe values before the start of the diet was 1448 µmol/L (STDV = 429), 23 (65.7%) were cPKU and 12 (34.3%) mPKU. 3d degree Parental consanguinity was found in 42.2% of studied families.
We have diagnosed 16 different alleles mutations, 5 mutations found in the seventh exon, 38/66 (75.6%) of the muted alleles was missense mutation, 22/66 (33.4%) was splice mutation and 6/66 (9%) was frame shift (Table 1), the most frequent mutations was p.R261Q which was found in 12.86% of the studied alleles and p.R243Q in 8.75% as well as p.F55>Lfs and IVS2+5G >C which They were found with the same frequency, while the IVS10-11G >A, p.P281L and p.R270K mutations were found with a relative frequency of 7.14% and IVS4+5G>T, p.S310F and p.P.E280 were found in 5.72%, and IVS9+5G>A had 4.29% While other mutations were found in less than 3% of the studied alleles.
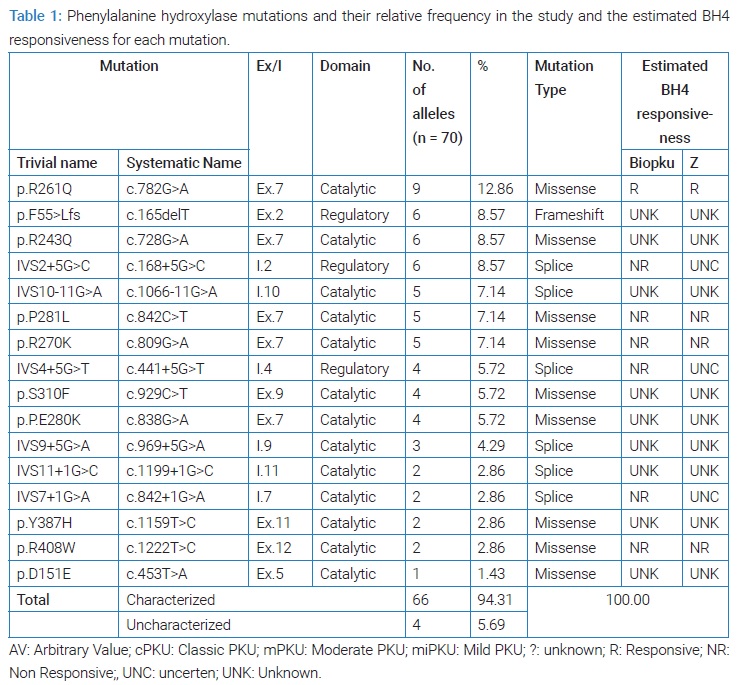
All 28/35 of the patients were homozygous. We found p.[R261Q];[R261Q] in four cases p.[R243Q];[R243Q] in three, p.[F55>Lfs];[p.F55>Lfs] in three and [IVS2+5G>C];[IVS2+5G>C] in three patients. 3/35 was compound heterozygote: [IVS4+5G>T];[IVS9+5G>A], [IVS4+5G>T];p.[R270K], p.[D151E]; [IVS10-11G>A] and 4/35 were heterozygote, they have a single mutant allele, two patients p.[R270K];[?] and one patient p.[R261Q];[?] and one patient p.[P281L];[?] and these cases needs furthermore studies [26] (Table 2).
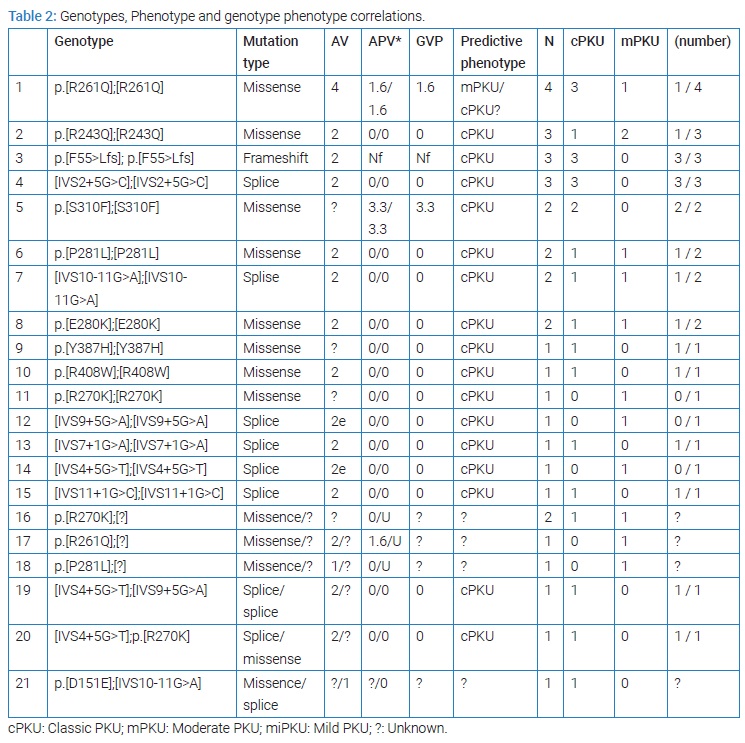
The results of the study of phenotypic compatibility with the genotype. Based on the data available in the databases in BioPKU db (last entry May 3, 2022) and based on the values of AVP for each mutant allele or the GVP of the sum of the two mutant alleles, we found that most of the mutations and genotypes encountered in our study were classified as cPKU-causing mutations (Table 2). The genotype allowed predicting phenotype in 17 homozygous genotypes encountered in 30 patients, including one homozygous genotype (p.R261Q) that was known to be associated with moderate form mPKU or cPKU [27]. All are classic mutations. 4 genotypes were not useful in predicting the phenotype, three of them are heterozygous and one is a heterozygote compound, where we did not find any information about the mutation p. [D151E], considering that it is a rare and unrecorded mutation (a new mutation) [24] (Table 3).
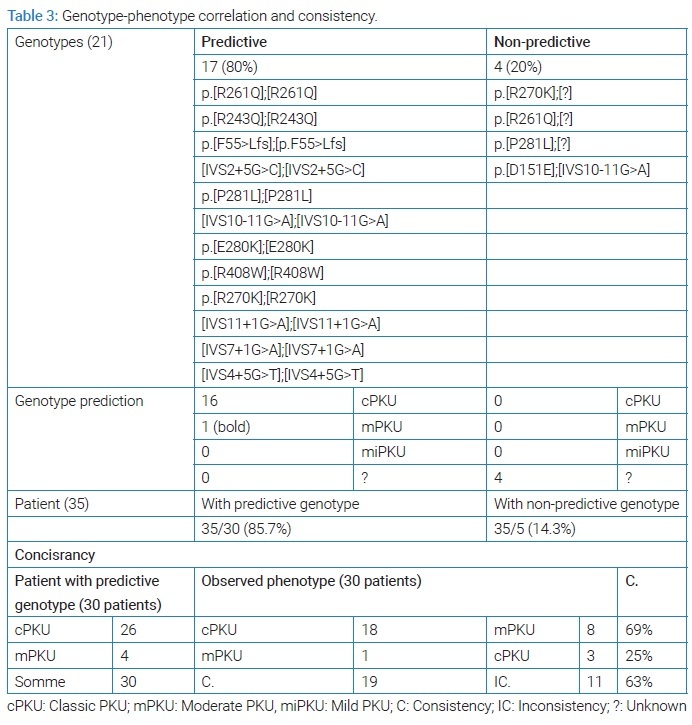
The homozygous mutations p.F55>Lfs, IVS2+5G>C, IVS7+1G>A, R408W, S310F, Y387H correlated with the occurrence of cPKU. The p.E280K, p.R243Q, p.P281L, IVS10-11G>A mutation correlated with cPKU in some cases and was associated with mPKU in others. p.R261Q correlated in one case to mPKU while 3 patients presented with cPKU. IVS9+5G>A R270K, IVS4+5G>T genotype were associated with mPKU.
IVS9+5G>A R270K, IVS4+5G>T genotype were associated with mPKU (Table 2). The three heterozygous compound cases [IVS4+5G>T];[IVS9+5G>A], [IVS4+5G>T];p.R270K], [p.[D151E];[IVS10-11G>A] associated with cPKU. Heterozygous cases p.[R261Q]];[?], p.[P281L]];[?] were associated with mPKU and p.[R270K]];[?] found in two cases, one cPKU and the other mPKU (Table 2). We can conclude that phenotype was predictable 30 patients (80.7%) (Table 3).
Predicting response to BH4 therapy
Based on the data available in the Biopku db databases and Zurfluh et al. (2008) [18], six mutations were described as Non-Responsive (NR) to BH4. we did not find enough information on response to BH4 therapy (UNK, UNC) for many of the common mutations encountered in our study such as p.F55>Lfs, p. R243Q and IVS2+5G>C (Table 1). Furthermore many of the mutations that before have been described as responsive to BH4 (R), including p.Y387H, IVS10-11G>A, IVS9+5G>A no comprehensive reports were received about It (UNK) or revealed NR or showed Uncertain Response (UNC) or Inconstant Response (IR) such in the p.R261Q mutant [27]. BioPKU db reported a response of the p.R261Q mutation to BH4 in 74% of the tested cases and a slow response in 7 out of the 33 patients studied carrying the homozygous IVS10-11G>A mutation and one patient out of two p.Y387H who was treated and did not respond Therefore, the response to BH4 can be expected in a few cases and it is for the last four phenotypes p.R261Q, IVS10-11G>A, which were occurred in only 8 patients in our study (22.9%), seven of whom showed cPKU and one patient had mPKU.
Discussion
This study shows a great diversity of genotypes for a small country such as Syria, where 16 different mutations were identified in 35 patients. This distinguishes the peoples of the Middle East in terms of the presence of a mixture of different races [28]. The p.R261Q mutation was the most frequent (12.86%), this mutation is commonly observed in the Mediterranean countries: 10.3% in Lebanon) 8.7% in Turkey, 8.5% in Sicily [29], 15.7% in southern Italy [30] and, IVS10-11G>A mutation was found in 7.1% of mutant alleles compared with the high prevalence of this mutation in Lebanon (27.6%) [29] and Turkey (24.6%) [31], and it is close to its incidence In Morocco and Tunisia [10,11]. The F55>Lfs mutation was found in 8.57% of the alleles, close to its incidence in Lebanon 7% [29] and it is mentioned that this mutation did not occur in other Arab countries, but only in the Arabs of Palestine [9]. The p.R270K mutation was It is found in 7.14% of alleles is commonly encountered in Europe and America, while it is rarely encountered in the Mediterranean region [32,33]. p.R408W, the most common mutation according to the BioPKU db, was only encountered in 2.86% of cases, we found IVS2+ 5G>C in 8.7% and p.R243Q by 8.7%. we found many different mutation in small percentages including p.P281L, IVS4+5G>T, IVS11+1G>C, IVS9+5G>A, p.E280K, they are common mutations in the Mediterranean region and Iran [34,35], p.S310F which was found in 5.72% was described in China, Korea [36,37] and Germany [38] p.Y387H that was found in 2.86% of our muted alleles was described in Germans [38], as well as in eastern Asia [37]. We found some rare mutations such as p.D151E and the database did not provide any information about it [39].
Study of phenotype-genotype correlation
Genotype-phenotype correlation was determinate for 35 patients with 21 different genotype, phenotype was predictable for 17 genotypes (80%) and unpredictable. 4 (20%) genotypes based on calculated AV (Guldberg et al.) [19], and the calculated APV and GPV according to BIOPKU (Table 3). It was possible to predict the phenotype based on the 17 genotypes in 30 patients: 26 cases expected to be associated with cPKU, only 18 of them were cPKU, and 4 expected to present mPKU only one case of them presented mPKU. Phenotype correlated with genotype in only 19 patient out of 30, we can conclude a consistency (phenotype genotype correlation) rate of 63% in our study (Table 3).
It’s known that the most severe disease is associated with the most severe mutations [3,6], the variation of the clinical phenotype is a reality and is related also to the diet [40], in a study of untreated families, it was found in the same family and for the same mutation a difference in the severity of the disease so that some of the diagnosed individuals are brothers of Severe disease and profound retardation appeared to be completely normal with a normal IQ, and the study concluded that there is no association between disease phenotype and the causative mutation in untreated children [41,42].
Since BH4-supplementation is not available in Syria, we made the first estimation of its potential benefit based on patients’ genotypes. Only few genotypes are estimated to be BH4 responsive this may due to the high allele frequency of severe PAH mutations. Even though, the BH4 responsiveness must be investigated by the response to the oral administration of BH4 (10 mg/kg to 20 mg/kg body weight) On all patients, especially with GVP more than 2.8 as APV values of 0, 5 or 10 ar considered to be strong predictors [25], and mutant compatible with mPKU or HPA [43]. It’s also indicated that mutations located in the catalytic domain were 68% of responsive mutations [18], We notice 13 PHA mutant alleles in the catalytic domain in our study (Table 1), and we had 9 patient with mPKU: one p.[R261Q];[R261Q], two p.[R243Q];[R243Q], one p.[P281L];[P281L], one [IVS10-11G>A];[IVS10-11G>A], one p.[E280K];[E280K], one p.[R270K];[R270K], one [IVS9+5G>A];[IVS9+5G>A], one [IVS4+5G>T];[IVS4+5G>T], one [IVS11+1G>C];[IVS11+1G>C], one p.[R270K];[?], one p.[R261Q];[?], one p.[P281L];[?] (Table 2). Those 9 patients must be studied; we notice some homozygote and heterozygote for R243Q to be BH4 responsive [44,45].
Conclusion
In Syria, the genetic spectrum showed a great diversity of mutation patterns, R261Q was the most frequent mutation in our studied patient (12.86%). This type is classified as a classic mutation, may be associated with mPKU or cPKU and may also respond to BH4. A trial to survey the response to BH4 therapy should be initiated in the future.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28(9):831–845.
- Williams RA, Mamotte CDS, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29(1):31–41.
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417–1427.
- Scriver CR, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly SW, Valle D, eds; Childs B, Kinzler KW, Vogelstein B, assoc eds. The Metabolic and Molecular Bases of Inherited Disease. Mc-Graw–Hill, New York. 2001;1667–1724.
- Lassker U, Zschocke J, Blau N, Santer R. Tetrahydrobiopterin responsivenesss in phenylketonuria. Two new cases and a review of molecular genetic findings. J Inherit Metab Dis. 2002;25(1):375–378.
- Scriver CR, Hurtubise M, Konecki D, Phommarinh M, Prevost L, Erlandsen H, et al. PAHdb 2003: what a locus-specific knowledgebase can do. Hum Mutat. 2003;21(4):333–344.
- Iraj Rezvani and Can H Ficicioglu. Phenylalanine, Defects in Metabolism of Amino Acids In: Kliegman RM, Stanton B, St J. Geme, Schor NF, editors. Nelson text book of pediatrics 20th ed. Philadelphia(PA): Elsiver Saunders. 2016;85:1:636–640.
- Effat LK, Essawi ML, Abd El Hamid MS, Hawari N, Gad YZ. Screening for six Mediterranean mutations in 90 Egyptian patients with phenylketonuria. Bratisl Lek Listy. 2008;109(1):17–19.
- Bercovich D, Elimelech A, Yardeni T, Korem S, Zlotogora J, Gal N, et al. A mutation analysis of the phenylalanine hydroxylase (PAH) gene in the Israeli population. Ann Hum Genet. 2008;72(Pt 3):305–309.
- Dahri S, Desviat LR, Perez B, Leal F, Ugarte M, Chabraoui L. Mutation analysis of phenylketonuria patients from Morocco: high prevalence of mutation G352fsdelG and detection of a novel mutation p.K85X. Clin Biochem. 2010;43(1–2):76–81.
- Khemir S, Siala H, Taieb SH, Cherif W, Azzouz H, Kéfi R, et al. Screening of three Mediterranean phenylketonuria mutations in Tunisian families. J Genet. 2012;91(1):91–94.
- Waters PJ, Parniak MA, Nowacki P, Scriver CR. In vitro expression analysis of mutations in phenylalanine hydroxylase: linking genotype to phenotype and structure to function. Hum Mutat. 1998;11(1):4–17.
- Walter JH, Lachmann RH, Burgard P. Hyperphenylalaninaemia in: Saudubray JM, van den Berghe G, Walter JH. Inborn Metabolic Diseases Diagnosis and Treatment 5th ed Springer- Verlag Berlin Heidelberg. 2012;17:253–264.
- Kure S, Hou DC, Ohura T, Iwamoto H, Suzuki S, Sugiyama N, et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr. 1999;135(3):375–378.
- Sterl E, Paul K, Paschke E, Zschocke J, Brunner-Krainz M, Windisch E, et al. Prevalence of tetrahydrobiopterine (BH4)-responsive alleles among Austrian patients with PAH deficiency: comprehensive results from molecular analysis in 147 patients. J Inherit Metab Dis. 2013;36(1):7–13.
- Rivera I, Mendes D, Afonso Â, Barroso M, Ramos R, Janeiro P, et al. Phenylalanine hydroxylase deficiency: molecular epidemiology and predictable BH4-responsiveness in South Portugal PKU patients. Mol Genet Metab. 2011:104 Suppl:S86–S92.
- Karacic I, Meili D, Sarnavka V, Heintz C, Thöny B, Ramadza DP, et al. Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiency. Mol Genet Metab. 2009;97(3):165–171.
- Zurfl€uh MR, Zschocke J, Lindner M, Feillet F, Chery C, Burlina A, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008;29(1):167–175.
- Guldberg P, Rey F, Zschocke J, Romano V, François B, Michiels L, et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet. 1998;63(1):71–79.
- Bercovich D, Elimelech A, Zlotogora J, Korem S, Yardeni T, Gal N, et al. Genotype–phenotype correlations analysis of mutations in the phenylalanine hydroxylase (PAH) gene. J Hum Genet. 2008;53(5):407–418.
- Daniele A, Scala I, Cardillo G, Pennino C, Ungaro C, Sibilio M, et al. Functional and structural characterization of novel mutations and genotype–phenotype correlation in 51 phenylalanine hydroxylase deficient families from Southern Italy. FEBS J. 2009;276(7): 2048–2059.
- Mallolas J, Vilaseca MA, Campistol J, Lambruschini N, Cambra FJ, Estivill X, et al. Mutational spectrum of phenylalanine hydroxylase deficiency in the population resident in Catalonia: genotype–phenotype correlation. Hum Genet. 1999;105(5):468–473.
- Santos LL, Fonseca CG, Starling ALP, Januário JN, Aguiar MJB, Peixoto MGCD, et al. Variations in genotype-phenotype correlations in phenylketonuria patients. Genet Mol Res. 2010;9(1):1–8.
- Garbade SF, Shen N, Himmelreich N, Haas D, Trefz FK, Hoffmann GF, et al. Allelic phenotype values: a model for genotype-based phenotype prediction in phenylketonuria. Genet Med. 2019;21(3):580–590.
- Vieira Neto E, Laranjeira F, Quelhas D, Ribeiro I, Seabra A, Mineiro N, et al. Genotype‐phenotype correlations and BH4 estimated responsiveness in patients with phenylketonuria from Rio de Janeiro, Southeast Brazil. Mol Genet Genomic Med. 2019;7(5):e610.
- Groselj U, Tansek MZ, Kovac J, Hovnik T, Podkrajsek KT, Battelino T. Five novel mutations and two large deletions in a population analysis of the phenylalanine hydroxylase gene. Mol Genet Metab. 2012;106(2):142–148.
- Gladys H, Alexander I, Bhattacharya K, Dennison B, Ellaway C, Thompson S, et al. The Molecular Bases of Phenylketonuria (PKU) in New South Wales, Australia: Mutation Profile and Correlation with Tetrahydrobiopterin (BH4) Responsiveness. JIMD Rep. 2014;14:55–65.
- Teebi SA. Genetic Diversity Among Arabs. In: Teebi, Ahmad (Ed.), Genetic disorders among Arab populations. Springer, Heidelberg, 2010;1:3–36.
- Karam PE, Alhamra RS, Nemer G, Usta J. Spectrum of mutations in Lebanese patients with phenylalanine hydroxylase deficiency. Gene. 2013;515(1):117–122.
- Daniele A, Cardillo G, Pennino C, Carbone MT, Scognamiglio D, Correra A, et al. Molecular epidemiology of phenylalanine hydroxylase deficiency in southern italy: a 96% detection rate with ten novel mutations. Ann Hum Genet. 2006;71(Pt 2):185–193.
- Dobrowolski SF, Heintz C, Miller T, Ellingson C, Ellingson C, Ozer I, et al. Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population. Mol Genet Metab. 2011;102(2):116–121.
- Eigel A, Dworniczak B, Kalaydjieva L, Horst J. A frameshift mutation in exon 2 of the phenylalanine hydroxylase gene linked to RFLP haplotype 1. Hum Genet. 1991;87(6):739–741.
- Guldberg P, Levy HL, Hanley WB, Koch R, Matalon R, Rouse BM, et al. Phenylalanine hydroxylase gene mutations in the United States: report from the Maternal PKU Collaborative Study. Am J Hum Genet. 1996;59(1):84–94.
- Baturina OA, Bondar AA, Tupikin AE, Zhabin SG, Morozov IV. [Mutation analysis of the phenylalanine hydroxylase gene of phenylketonuria patients of Kemerovskaya Oblast' and Saha Republic]. Tsitol Genet. 2012;46(4):40–47.
- Hamzehloei T, Hosseini SA, Vakili R, Mojarad M. Mutation spectrum of the PAH gene in the PKU patients from Khorasan Razavi province of Iran. Gene. 2012;506(1):230–232.
- Nana Li, Jia H, Liu Z, Tao J, Chen S, Li X, et al. Molecular characterisation of phenylketonuria in a Chinese mainland population using next-generation sequencing. Sci Rep. 2015;5:15769.
- Lee DH, Koo SK, Lee KS, Yeon YJ, Oh HJ, Kim SW, et al. Jung The molecular basis of phenylketonuria in Koreans. J Hum Genet. 2004;49(11):617–621.
- Eisensmith RC, Goltsov AA, O'Neill C, Tyfield LA, Schwartz EI, Kuzmin AI, et al. Recurrence of the R408W mutation in the phenylalanine hydroxylase locus in Europeans. Am J Hum Genet. 1995;56(1):278–286.
- Bonafé L, Blau N, Burlina AP, Romstad A, Güttler F, Burlina AB. Treatable neurotransmitter deficiency in mild phenylketonuria. Neurology. 2001;57(5):908–911.
- Kayaalp E, Treacy E, Waters PJ, Byck S, Nowacki P, Scriver CR. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype–phenotype correlations. Am J Hum Genet. 1997;61(6):1309–1317.
- Ramus SJ, Forrest SM, Pitt DB, Saleeba JA, Cotton RG. Comparison of genotype and intellectual phenotype in untreated PKU patients. J Med Genet. 1993;30(5):401–405.
- Mitchell JJ. Phenylalanine Hydroxylase Deficiency. 2000 Jan 10 [Updated 2013 Jan 31]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2017;1993–2016.
- Quirk EM, Dobrowolski SF, Nelson BE, Coffee B, Singh RH. Utility of phenylalanine hydroxylase genotype for tetrahydrobiopterin responsiveness classification in patients with phenylketonuria. Mol Genet Metab. 2012;107(1–2):31–36.
- Tao J, Li N, Jia H, Liu Z, Li X, Song J, et al. Correlation between genotype and the tetrahydrobiopterin-responsive phenotype in Chinese patients with phenylketonuria. Pediatr Res. 2015;78(6):691–699.
- Zhu T, Ye J, Han L, Qiu W, Zhang H, Liang L, et al. The predictive value of genetic analyses in the diagnosis of tetrahydrobiopterin (bh4)-responsiveness in chinese phenylalanine hydroxylase deficiency patients. Sci Rep. 2017;7(1):6762.
Keywords
Phenylketonuria; Genotype; Phenotype; Response to BH4; Syria
Cite this article
Alasmar D. Predicting biopterin responsive cases by studying phenylalanine hydroxylase mutations in children diagnosed with phenylketonuria in Syria. Glob J Pedia. 2022;1(2):1–8.
Copyright
© 2022 Diana Alasmar. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
