Pyruvate Dehydrogenase Deficiency: Rare Mutation of the E1ß Subunit Gene
* Haddad Samir;
Rym A;
Zeineb E;
Sabrine B;
Rahma C;
Dorra F;
Nizar N;
-
* Haddad Samir: Department of Pediatrics A, Béchir Hamza Children's Hospital, Tunis, Tunisia.
-
Rym A: Department of Pediatrics A, Béchir Hamza Children's Hospital, Tunis, Tunisia.
-
Zeineb E: Department of Pediatrics, Tunis Pediatric Clinic, Tunisia.
-
Sabrine B: Department of Pediatrics, Tunis Pediatric Clinic, Tunisia.
-
Rahma C: Department of Pediatrics, Tunis Pediatric Clinic, Tunisia.
-
Dorra F: Department of Pediatrics, Tunis Pediatric Clinic, Tunisia.
-
Nizar N: Department of Pediatrics, Tunis Pediatric Clinic, Tunisia.
-
Sep 21, 2022 |
-
Volume: 1 |
-
Issue: 2 |
-
Views: 2355 |
-
Downloads: 1888 |
Abstract
Pyruvate Dehydrogenase Complex (PDC) defects is a far-famed cause of metabolic disorders. Progressive neurological symptoms usually start in infancy, but may be evident at birth or in later childhood. They can induce delayed psychomotor development, brain malformations, hypotonia, seizures, ataxia, West or Leigh-like syndrome. PDC E1β deficiency has been described in only seven patients.
In this report, we add a novel case of PDC E1β deficiency with different clinical picture than previously reported cases. Index cases have later age of onset and initial symptoms mimicking encephalitis.
Introduction
The Pyruvate Dehydrogenase Complex (PDC) is one of the key enzymes in pathways of energy production and biosynthesis of lipids from glucose-derived carbons [1]. It converts pyruvate into acetyl-coA. This process is called pyruvate decarboxylation. Acetyl-CoA can then be used in the citric acid cycle to effect cellular respiration, and this complex links the metabolic pathway of glycolys is to the citric acid cycle. Hence the production of cellular ATP is reduced in cases of PDC deficiency [2,3].
Neuromuscular and neurological degeneration are the most common damages despite the phenotypic variability of PDC deficiency. Neurological involvement usually begins in early childhood. Its appearance in adult hood is very rare. They can induce delayed psychomotor development, brain malformations, hypotonia, seizures, ataxia, West or Leigh-like syndrome [1–4].
PDC is a complex of three enzymes: pyruvate dehydrogenase (E1), dihydrolipoamide transacetylase (E2), and dihydrolipoamide dehydrogenase (E3). The E1 enzyme has a structure composed of two α and two β subunits and has a thiamine pyrophosphate binding site. The proteins constituting the enzyme complex are coded for by nuclear genes and the disorder is inherited through both X-linked and autosomal recessive modes of inheritance [4].
The majority of cases of PDC deficiency are due to mutations in the PDHA1 gene which encodes for the E1α subunit. A few cases are attributable to mutations in the genes for E3, E3 binding protein, E1-phosphatase and E2 [5].
In previous studies, E1β deficiency was described in only seven patients. Mutations in the PDC gene were reported in six of them. Most of these PDC E1β deficiency cases are relatively severe and did not survive as long as those with E1α or E3β P deficiency [6].
Here in, we add a novel case of PDC E1β deficiency with different clinical picture than previously reported cases. Index case has later age of onset and initial symptoms mimicking encephalitis.
Case Report
An 8-year-old female born at term to consanguineous parents was admitted for management of consciousness disorders. She is the second child of healthy parents with a healthy 12-year-old sister. Family history of neurodegenerative disorders was denied. Her neuro developments: motor, language and cognitive development were normal. She had good academic results.
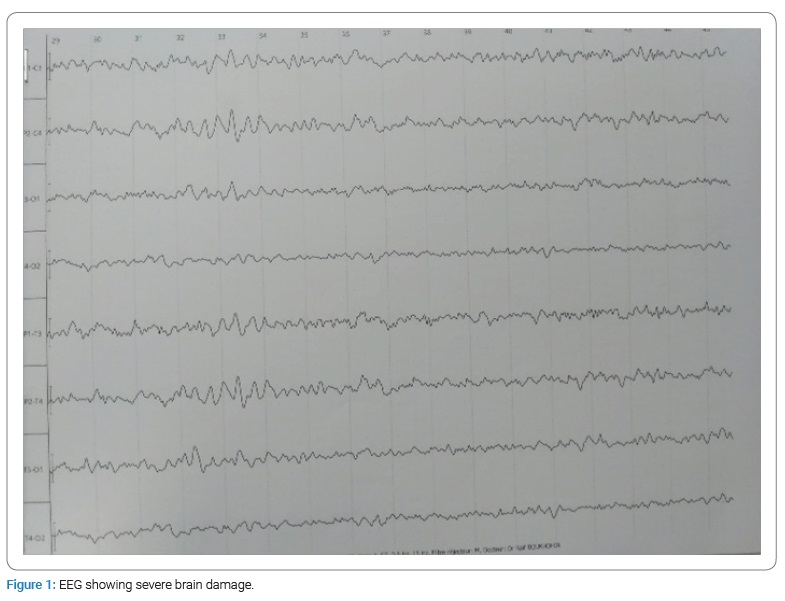
Two weeks before admission, she had fever with vomiting and cough treated symptomatically, then gradual onset of gait disturbances followed by rapidly worsening consciousness. On examination, we found an unconscious patient with a Glasgow Coma Scale score of 7. She was quickly incubated and put on mechanical ventilation. In front of this clinical picture, we suspected encephalitis that was treated with Zovirax, cefotaxime and vancomycin. Lumbar puncture was normal. Further tests were requested on her Cerebrospinal Fluid (CSF) including multiplex polymerase chain reaction, anti-NMDAR, anti-MOG, anti-GAD and anti-AMPA 2 antibodies. But there were negative. Viral serologies: Herpes simplex virus 1 and 2 Epstein-Barr virus and cytomegalovirus were also negative. Electroencephalogram (EEG) showed severe brain damage (Figure 1). Initial brain MRI was compatible with acute disseminated encephalomyelitis with extensive active lesions above and below the tentorium (Figure 2). She received IV methylprednisolone boli for 5 days then venoglobulin 1 g/kg per day for two days without clinical improvement. Then, she developed seizures that were treated with sodium valproate and levetiracetam.

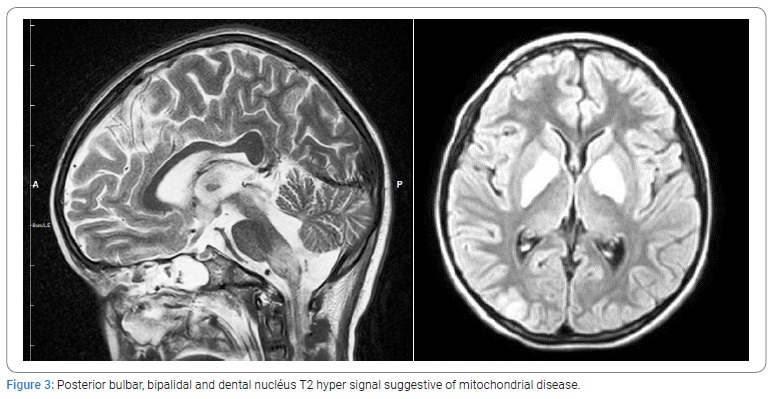
A control brain MRI performed after one week showed lesions suggestive of mitochondrial disease in its acute phase (Figure 3). Biochemical studies showed metabolic acidosis, ammonia was elevated at 152 µmol and high level of CSF lactate at 2.01 mmol. Urine Organic Acid analysis and chromatography of aminoacid in blood and urine were normal.
Whole-Exome Sequencing was performed and we identified a homozygous pathogenic variant in the PDHB gene: c.313 A > G p. (Met101 Val). The result is consistent with a genetic diagnosis of autosomal recessive pyruvate dehydrogenase E1-beta deficiency. The genetic study has not been carried out for the rest of the family.
We noted a slight improvement after one month of evolution on ketogenic diet and vitamin B1: progressive awakening, spontaneous opening of the eyes with ocular pursuit, weak motor reactions to stimulation with resumption of spontaneous but ineffective breathing.
Discussion
We present a rare case of PDC E1β deficiency. Whole-Exome Sequencing identified a homozygous pathogenic variant in the PDHB gene: c.313 A > G p. (Met101 Val). This variant has previously been described by Quintana et al. [6] and Asencio et al. [7].
Human PDC is a multienzyme complex of 6 subunits, pyruvate dehydrogenase (E1), dihydrolipoyl acetyltransferase (E2), dihydrolipoyl dehydrogenase (E3), an E3-binding protein (E3BP), and two dedicated regulatory components—Pyruvate Dehydrogenase Kinase (PDK) and Pyruvate Dehydrogenase Phosphatase (PDP) [4–8].
E1β subunit of PDC has been mapped to 3p13-q23. E1β mutations are still rare and account for only a minority of cases of PDC deficiency, they currently appear to be the fourth most common documented cause of PDC deficiency. The cases of E1β deficiency previously published constitute a total of seven cases [6,9].
Most cases of PDC E1β deficiency are relatively severe and did not survive as long as E those with E1α or E3BP deficiency. They have fairly similar features to E1α deficiency, predominantly impairment of neurological and motor function, including particularly developmental delay, hypotonia, structural abnormalities of the brain, particularly ventriculomegaly, colossal dysgenesis and findings consistent with Leigh syndrome; hyperlactatemia; and a circulating lactate: pyruvate ratio ≤ 20 [1,4,6,9].
Inhibition of PDC activity impairs the mitochondrial oxidation of pyruvate and promotes its cytoplasmic reduction to lactate or transamination to alanine. Diminished flux through the PDC alsodecreases the availability of acetyl CoA for the Citric Acid Cycle (CAC). In turn, reduction of both PDC and CAC activities decreases the generation of reducing equivalents (as NADH and FADH2) that donate electrons to the respiratory chain to complete the process of oxidative phosphorylation. Consequently, potentially any congenital or acquired defect in any PDC component may give rise to lactic acidosis and to cellular energy failure, the latter most commonly expressed as progressive neurological and neuromuscular deterioration [2,9–10].
Never the less, the clinical spectrum of PDC deficiency is broad, ranging from neonatal death with over whelming lactic acidosis to a relatively benign course early in life with few overt signs [9]. Index case was a symptomatic with good psychomotor development until the age of 8 years. Diagnostic difficulties were that the patient has no pathological history, initial symptoms mimicking encephalitis and an initial brain MRI evoking acute disseminated encephalomyelitis. Secondarily, diagnosis was suspected in view of the neurological impairment: no improvement under treatment even aggravation with seizures, lactic acidosis and a control cerebral MRI evoking a mitochondrial disease in its acute phase. The diagnostic confirmation was made by the genetic study.
Our patient showed a slight improvement after one month on a ketogenic diet and vitamin B1. Indeed ketogenic diet leads to an increasein circulating plasma levels of free fattyacids and ketone bodiesprovidingalternatefuel for brain metabolism and also resulting in a reduction in blood and intra cellular levels of lactate and pyruvate due to the reduction or removal of dietary carbohydrates [1–11]. Sofou et al. [11], realized the first longitudinal study on long-term efficacy and safety outcomes of Ketogenic Diet (KD) in 19 pediatric patients with PDC deficiency. They showed in this study that blood lactate decreased significantly after KD initiation. This explains the positive and safe effect of KD mainly in the areas of epilepsy, ataxia, sleep disturbance, speech/language development, social functioning, and frequency of hospitalizations. Poor dietary compliance was associated with relapsing ataxia and stagnation of motor and neurocognitive development [11]. Pliss et al. [1], showed that early prenatal treatment with a ketogenic diet might be very beneficial during pregnancy with a family history of PDC deficiency (with a previously affected sibling) or with a positive prenatal diagnosis of PDC deficiency), because this dietary treatment has the potential to reduce irreversible cerebral damage occurring during the prenatal period.
Several studies have shown that supplementation with high doses of thiamine (vitamin B1) is beneficial in meeting the increased Km requirement for thiamine pyrophosphate associated with certain PDHA1 mutations and/or for enzyme stability, as well as the administration of dichloroacetate which is known to inhibit PDH kinase decreased blood cerebrospinal fluid lactate concentrations in a large number of PDC deficient children [1,12].
Conclusion
The clinical spectrum of PDC deficiency is broad, the dominant clinical phenotype includes neurological and neuromuscular degeneration.
The present study reports a novel case carrying the mutation c.313 A > G p. (Met101 Val) with different clinical picture of PDC E1β deficiency. Index case showed a slight improvement on ketogenic diet and vitamin B1.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent from the family was obtained for this publication.
References
- Pliss L, Jatania U, Patel MS. Beneficial effect of feeding a ketogenic diet to mothers on brain development in their progeny with a murine model of pyruvate dehydrogenase complex deficiency. Molecular Mol Genet Metab Rep. 2016;7:78–86.
- Bhandary S, Aguan K. Pyruvate dehydrogenase complex deficiency and its relationship with epilepsy frequency--An overview. Epilepsy Res. 2015;116:40–52.
- Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236–250.
- Ma Y, Zhang Y, Zhang T, Man Z, Su XM, Hao SJ, et al. Pyruvate dehydrogenase deficiency disease detected by the enzyme activity of peripheral leukocytes. Mol Genet Genomic Med. 2021;9(8):e1728.
- Okajima K, Korotchkina LG, Prasad C, Rupar T, Phillips JA 3rd, Ficicioglu C, et al. Mutations of the E1beta subunit gene (PDHB) in four families with pyruvate dehydrogenase deficiency. Mol Genet Metab. 2008;93(4):371–380.
- Quintana E, Mayr JA, García Silva MT, Font A, Tortoledo MA, Moliner S, et al. PDH E1β deficiency with novel mutations in two patients with Leigh syndrome. J Inherit Metab Dis. 2009;32(Suppl 1):S339–S343.
- Asencio C, Rodríguez-Hernandez MA, Briones P, Montoya J, Cortés A, Emperador S, et al. Severe encephalopathy associated to pyruvate dehydrogenase mutations and unbalanced coenzyme Q10 content. Eur J Hum Genet. 2016;24(3):367–372.
- Prasad C, Rupar T, Prasad AN. Pyruvate dehydrogenase deficiency and epilepsy. Brain Dev. 2011;33(10):856–865.
- Patel KP, O’Brienb TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;105(1):34–43.
- Bedoyan JK, Hecht L, Zhang S, Tarrant S, Bergin A, Demirbas D, et al. A novel null mutation in the pyruvate dehydrogenase phosphatase catalytic subunit gene (PDP1) causing pyruvate dehydrogenase complex deficiency. JIMD Rep. 2019;48(1):26–35.
- Sofou K, Dahlin M, Hallböök T, Lindefeldt M, Viggedal G, Darin N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J Inherit Metab Dis. 2017;40(2):237–245.
- Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Ogawa Y, et al. Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim Biophys Acta. 2002;1588(1):79–84.
Keywords
Pyruvate dehydrogenase complex deficiency; PDC E1β; Metabolic acidosis; Whole-exome sequencing; Ketogenic diet
Cite this article
Samir H, Rym A, Zeineb E, Sabrine B, Rahma C, Dorra F, et al. Pyruvate dehydrogenase deficiency: rare mutation of the E1β subunit gene. Glob J Pedia. 2022;1(2):1–5.
Copyright
© 2022 Haddad Samir. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
