Abstract
Guillain-Barré Syndrome (GBS) is an uncommon autoimmune disorder characterized by polyradiculoneuropathy. This dramatic syndrome in otherwise healthy individuals imposes significant fear and anxiety on the patient and the family alike. We present a case of a 8-year-old girl with acute onset diplopia followed by predominant proximal muscle weakness in all four limbs. She had no prior history of travel, infection or immunization. Clinical semiology was suggestive of polyneuropathy, however electrophysiological investigations were not contributory. Lumbar puncture and anti-ganglioside serology were consistent with the diagnosis of GBS. The treatment by Intravenous Immunoglobulin (IVIG) needs to be instituted in a timely manner to prevent dreaded respiratory complications. GBS should be viewed in clinical spectrum and we emphasize role of anti-ganglioside antibodies in documentation, parental counselling and treatment of GBS.
Introduction
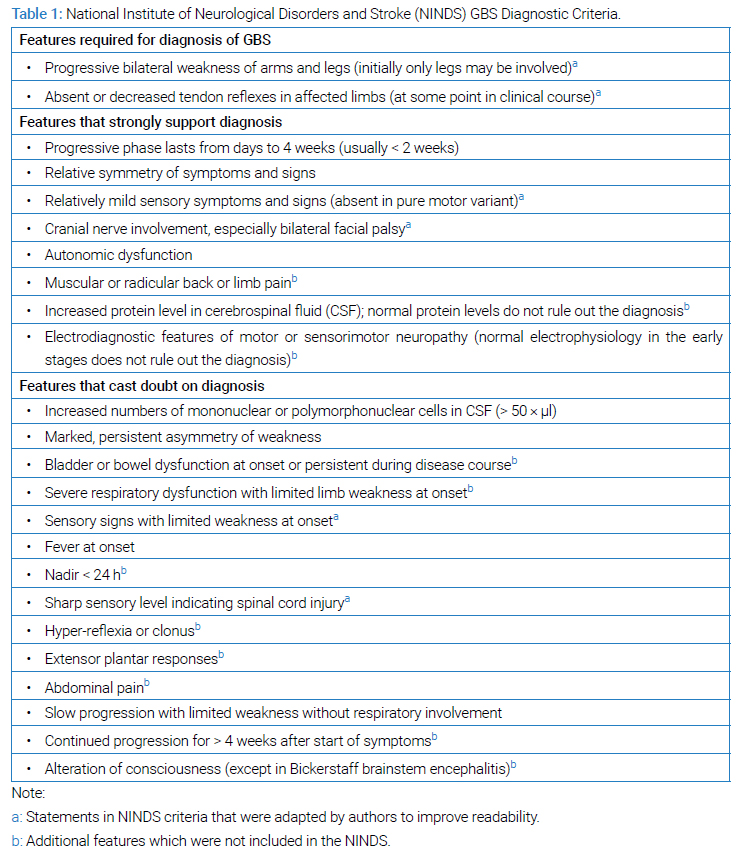
Inability to bear weight is not an uncommon presentation in paediatric clinical practice. Post-Polio eradication, worldwide leading cause of symmetrical acute flaccid paralysis is Guillain–Barré Syndrome (GBS). GBSclinically presents with progressive ascending weakness and diminished or absent reflexes with an underlying pathophysiology of acute autoimmune polyradiculoneuropathy [1]. The Brighton criteria are used to assist in the diagnosis of GBS and help distinguish between low-risk and high-risk patients. The criteria includes – Absence of alternative diagnosis for weakness, Diminished or absent deep tendon reflex in weak limbs, Monophasic course and time between onset and nadir, Bilateral and flaccid weakness of limbs, CSF cell counts < 50 cell/uL, CSF protein concentration > normal value and NCS findings consistent with one of the subtypes of GBS. Commonly, a National Institute of Neurological Disorders and Stroke (NINDS) criterion is used to determine the presence of GBS (Table 1).
An important variant of GBS is Miller Fisher Syndrome which is characterized by weakness of eye muscles (ophthalmoplegia), impaired limb coordination (ataxia) and absent tendon reflexes (areflexia). The diagnosis has traditionally relied on clinical findings, Cerebrospinal Fluid (CSF) analysis (high protein, low cell counts, lymphocytic predominance), Nerve Conduction Velocity (NCV) studies (motor and/or sensory nerve conduction slowing or block, prolonged distal motor latency and prolonged or absent F-wave), and sometimes imaging of spine (enhancement of anterior spinal nerve roots) and brain (transient involvement of brainstem and basal ganglia by high T2 signal and some minor restricted diffusion in Bickerstaff encephalitis). Treatment involves the administration of IVIG, plasma exchange and supportive care. Early diagnosis and management has shown to carry better prognosis in terms of fewer and milder neurological deficits. Further, early treatment diminishes the probability of ventilatory support requirement in the acute phase of the disease [2,3].
Notably, a sizable proportion of GBS cases demonstrate overlapping features leading to diagnostic dilemma [4]. The spectrum of features may occur in isolation or in combination of – proximal muscle group involvement, normal nerve conduction studies, normal imaging, dysautonomia, asymmetrical areflexia, truncal ataxia, facial weakness, mood irritability, sensory predominance, headache, neck rigidity, persistent bowel-bladder involvement, ophthalmoplegia, ultra-rapid progression and varying rate of recovery. In such scenario, utility of specialised serological tests come to the fore. Various ganglioside antibodies have featured in literature with their own phenotypic characteristics aiding in diagnosis, confident management and prognostication [5].
Case Presentation
An 8-year-old girl presented to the ER with acute onset double vision for 3 days and inability to bear weight for 2 days. Since morning, she could not move from supine to prone position or lying down to sitting up position without significant assistance. There was no history of recent travel, respiratory or gastrointestinal symptoms before her presentation. Her medical history is insignificant with lack of any noteworthy events, chronic illness, and serious illness or drug allergies. Her physical examination revealed normal vital signs. There was no hypertension, postural drop in blood pressure, tachycardia or arrhythmia suggesting autonomic nervous system sufficiency. Classical history of “light in the morning and heavy in the evening” was not reported. Neurological examination findings were consistent with a flaccid-type quadriparesis of trunk, upper limbs and lower limbs. In all four limbs, muscle tone was reduced, diminished deep tendon reflexes in bilateral knees and absent plantar response. In the upper limbs, proximal muscle group power was 2/5 on the right and 3/5 on the left, distal muscle group power was 4/5 bilaterally.
In the lower limb examination, proximal power was 2/5 and distal 4/5 bilaterally. She had paraesthesia but no neuropathic pain in both the lower limbs. Her facial diplegia (left more than right) was appreciable, she could not squeeze her eyes shut completely or blow up the cheeks while trying to push the air out against pursed lips efficiently. Her lateral gaze was severely restricted and upward gaze was also restricted bilaterally, hinting towards Miller Fisher syndrome. No headache, confusion, decreased alertness, abnormal movements or lack of comprehension was reported pointing away from Bickerstaff Encephalitis. Gait could not be assessed due to severe weakness and no cerebellar signs were absent. Other systemic examination was unremarkable.
Investigations
On day 1 of hospitalization and day 4 of illnessroutine tests including blood glucose, full blood counts, C reactive protein, creatinine, urea, electrolytes, calcium, magnesium, phosphate, liver function tests, Creatine Phosphokinase (CPK), vitamin B12 and vitamin D were within normal limits. ECG showed normal sinus rhythm and COVID-19 PCR test for SARS-CoV-2 RNA came back negative as a part of routine hospital admission screen. In view of diplopia and predominant cranial nerve involvement, a non-contrast MRI of brain and whole spine was performed to rule out pathologies explaining the clinical presentation. An incidental finding of a plaque in the vicinity of the left frontal horn was seen while no spinal cord pathology was noted. There was no evidence of spinal cord compression, transverse myelitis, spinal cord infarction or spinal stenosis.Ice pack test and neostigmine test couldn’t be carried out because of parental objections.
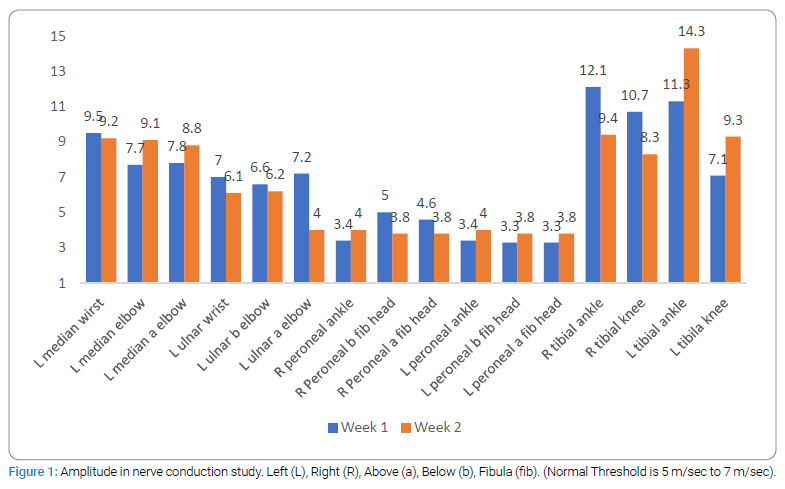
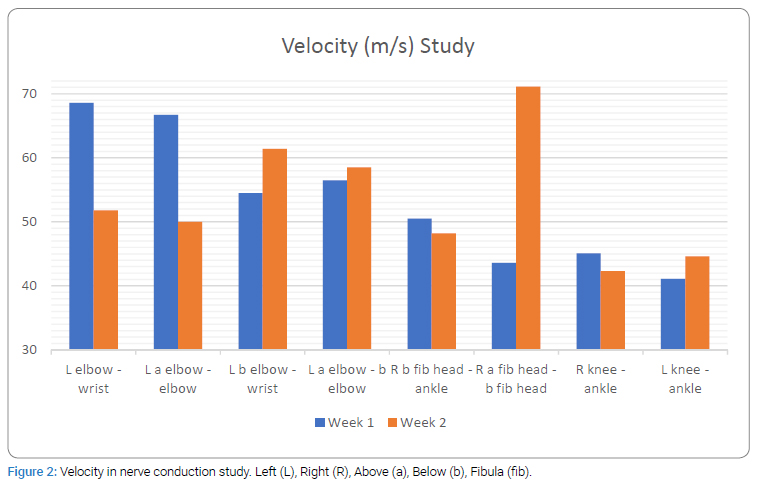
Consideringflaccid paralysis suggeting GBS, and significant ocular involvement prompting a possibility of neuromuscluar junction pathology such as myasthenia gravis, electrophysiological testing was carried out. In both, first and second week of hospitalization, patient’s nerve condiction study (Figure 1 and 2), Repetitive Nerve Stimulation Test (RNST) and bink reflex were normal. In the secnond week of illness CSF examination revealed raised proteins (142, range 15 mg/dl to 60 mg/dl), slighltlly raised cell counts (15/cmm) with 90 percent lymphocytic predominance. Towards the third week of illness, the IgG antiganglioside antibody against GD1a was found to be positive (consistent with the diagnosis of GBS) while IgG antibodies against GM1, GM2, GM3, GD1b, GT1b and GQ1b were negative. IgM antibodies against the all the above gangliosides were negative.
Differential Diagnosis
Given the initial presentation of diplopia and facial muscle weakness, neuromuscular junction conditions such as Myasthenia gravis was one of the considerations. However, acute flaccid polyneuropathy strongly suggested GBS. In view of a sudden, non-progressive and predominantly proximal muscle weakness, viral myositis was ruled out by normal CPK. Spinal cord compression from spinal cord infarcts, transverse myelitis or spinal stenosis needed to be ruled out. Normal MRI spine, absence of spastic paraparesis, bowel or bladder involvement and the absence of back pain made spinal cord compression unlikely.
Peripheral neuropathy can present as flaccid paraparesis or quadriparesis, but usually follows a more gradual course unlike the presentation of this girl. Normal ANA, negative ANCA, normal complement C3 and C4 levels ruled out the possibility of other autoimmune disorders or vasculitis causing flaccid neuropathy. There was no travel history to regions with polio or tick endemicity, which can closely mimic GBS. Albumin-cytological dissociation and positive anti-GD1a IgG antibody confirmed the diagnosis of GBS.
Treatment
Based on clinical grounds, on day 3 of hospitalization and day 6 of illness, the patient was commenced on IVIG at a dose of 2 g/kg body weight divided over 4 consecutive days, 72 hours after IVIG commencement, patient developed low grade fever for paracetamol was given orally and IVIG infusion rate was tapered to complete the total dose in 5 days time. Inflammatory markers were all within normal range, therefore antibiotic cover upgradation was not required. Neuropathic pain was not reported during the course of hospitalization and Gabapentin was not used. In absence of respiratory complications patient did not require oxygen or ventilator support.
Outcome and follow-up
At discharge, on day 14 of hospitalization and day 17 of illness, she was able to sit up from supine position with support. Her power in both upper and lower limb proximal muscle groups improved significantly, but remained weak in contrast to distal muscle groups which achieved 5/5 power bilaterally. Her facial muscles seemed more powerful, ophthalmoplegia was not profound and diplopia was not reported (Figure 3). Her gait was wide based and unsteady but did not require support.

Discussion
Several auto-ganglioside antibodies causing autoimmune neuropathy have been studied in the last couple of decades. Anti-GD1a antibody has emerged as an important marker of Acute Motor Axonal Neuropathy (AMAN). It is related to severe axonal degeneration of peripheral nerves with increased likelihood of patients requiring mechanical ventilation [6,7]. Overwhelming majority of IgG anti-GD1 antibody related clinical reports have been described in adult population and pediatric literature is scarce. In adults, GD1a antibody presentation is a close replication of classical GBS progression of gradual ascending flaccid paralysis with relatively early recovery. Patients with positive IgG anti-GD1a antibodies, presenting with isolated cranial neuropathy without limb weakness have also been reported [8]. A retrospective study of 33 anti-GD1a positive patients reported a strong association with pure motor and axonal variants of GBS [9]. In another prospective study of 138 patients, it was found that anti-GD1a antibody phenotype was not associated with Miller Fisher Syndrome [10]. Also, isolated facial diplegia with distinctly positive anti-GD1a antibodies is exceedingly rare [11]. In another study which recruited 8600 patients to study GBS variant with facial diplegia, none were associated with anti-GD1a IgG and only anti-GM2 IgG antibody was found to be associated with facial involvement in 5 patients [12].
Both GBS and MFS are characterized by a progressive weakness that typically starts in the legs and spreads to the arms and upper body. Common features seen in both the conditions include sensory symptoms (such as tingling, numbness, or pain in the limbs), diminished or absent deep tendon reflexes, respiratory and cardiovascular complications in severe cases, triggered by infection [1]. The overlapping presentation poses a challenge in diagnostic determination.
To the best of our knowledge, this is the first pediatric report of patient with anti-GD1a IgG positive antibody with ophthalmoplegia and proximal muscles involvement. The natural history in this case depicted acute onset illness with progressive aggravation suggesting infectious or immune inflammation. Relative sparing of distal limbs and absence of typical ascending disease progression points away from typical GBS. On the other hand, cranial nerve involvement as primary complaint without areflexia or ataxia suggests an incomplete MFS. This is an interesting and rare pattern which can create significant diagnostic dilemma. Awareness of such possible GBS patterns is necessary for comprehensive and effective medical management. Lack of typical features of GBS with uncharacteristic normal electrophysiological and imaging findings cause additional challenge in parental counselling. At the outset of suspicion of polyneuropathy related to GBS, parents must be made aware that investigations need to be carried out for a definitive diagnosis but normalcy of results cannot halt treatment of GBS.
Parent’s perspective
GBS is not a condition that people normally know about. It is frightening to see your child not able to see, sit or walk all of a sudden. Starting the treatment seemed the most important thing to do but none of the imaging or nerve tests were able to determine if it was GBS. This creates confusion and doubt. It is the uncertainty about the disease, chances of recovery and chances of it happening again, which is the most difficult thing to confront.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
Keywords
GBS; Neuropathy; Antibody; Anti-GD1a
Cite this article
Shanker V, Singh PK. Anti-Gd1a Positive Auto Antibodies Presenting with Guillain–Barré Syndrome and Miller Fisher Syndrome Overlap. Ann Neur Res Stud. 2023;2(1):1–6.
Copyright
© 2023 Varnit Shanker. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
