Three Siblings with Aarskog Scott Syndrome Together with Mental Retardation and Giant Megacolon
* Hisashi Kawashima;
Nishimata S;
Shinji S;
Morishima Y;
Tsutsumi N;
Kashiwagi K;
Amano T;
Tamura M;
Ayabe S;
Nakashima K;
-
* Hisashi Kawashima: Department of Pediatrics, Kohseichuo Hospital, Tokyo, Japan; Department of Pediatrics and Adolescent Medicine, Tokyo Medical University, Tokyo, Japan.
-
Nishimata S: Department of Pediatrics and Adolescent Medicine, Tokyo Medical University, Tokyo, Japan.
-
Shinji S: Department of Pediatrics and Adolescent Medicine, Tokyo Medical University, Tokyo, Japan.
-
Morishima Y: Department of Pediatrics and Adolescent Medicine, Tokyo Medical University, Tokyo, Japan.
-
Tsutsumi N: Department of Pediatrics and Adolescent Medicine, Tokyo Medical University, Tokyo, Japan.
-
Kashiwagi K: Department of Pediatrics and Adolescent Medicine, Tokyo Medical University, Tokyo, Japan.
-
Amano T: Next Generation Human Disease Model Team, RIKEN BioResource Research Center, Japan.
-
Tamura M: Technology and Development Team for Mouse Phenotype Analysis, RIKEN BioResource Research Center, Japan.
-
Ayabe S: Experimental Animal Division, RIKEN BioResource Research Center, Japan.
-
Nakashima K: Gene Engineering Division, RIKEN BioResource Research Center, Japan.
-
Feb 26, 2024 |
-
Volume: 2 |
-
Issue: 1 |
-
Views: 1430 |
-
Downloads: 1478 |
Abstract
We found a new candidate disease-causing gene in 3 siblings who have Aarskog-Scott syndrome together with mental retardation and megacolon. Using exome analysis, ZFHX3, which has been reported to be associated with Hirschsprung disease, was identified as a candidate causative gene for their disease (mutations c.G756A:p.A2521T and c.G4428A:p.M1476I). In silico analysis suggested that A2521T was a functionally damaging mutation. Mice with a frame shift mutation in ZFHX3 were fetal lethal. Mice with the A2530T (which corresponds to human A2521T) knockout mutation in ZFHX3 were viable and did not have any obvious neurological abnormalities.
Introduction
Aarskog Scott Syndrome (AAS) is an inherited disease showing either X-linked recessive or autosomal-dominant inheritance (OMIM #305400) [1], and is known to be caused by abnormalities in the FGD1 gene [2]. However, mutations in the FGD1 gene have been found in only 18.3% of AAS patients, and other causative genes have remained unknown to date [3]. The disease is accompanied with skeletal dysplasia, urinary tract anomalies, telecanthus, external ear malformation, maxillary hypoplasia, and abnormalities of the sigmoid colon [4,5]. We present 3 siblings with AAS, who have chronic constipation with megacolon and psychomotor developmental retardation. The patients were found to have ZFHX3 gene variants. ZFHX3 is also called as Zinc finger homeobox protein 3 and encodes a transcription factor with multiple homeodomains and zinc finger motifs, and regulates myogenic and neuronal differentiation. However, as the patients also had mental retardation and giant megacolon, our data indicates that ZFHX3 is not an independent candidate causative gene for AAS, and the possibility of clinical complications should be taken into consideration.
Case Presentation
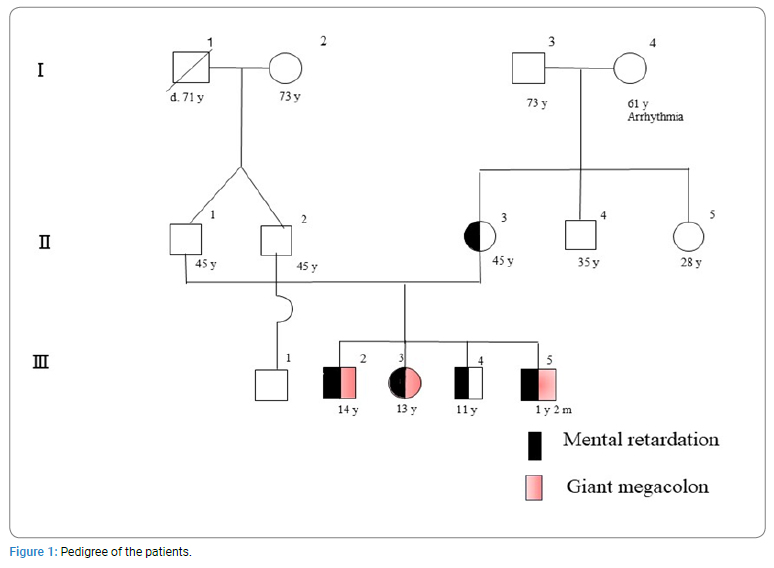
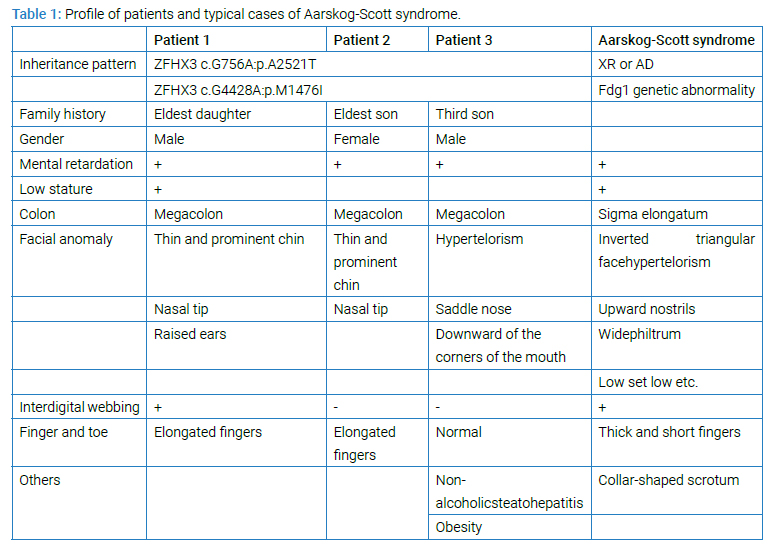
The sister (middle of 4 siblings; (Figure 1) complained of intractable constipation and abdominal tension, and was admitted to our department at the age of 8 years. She had no particular past medical history except for psychomotor developmental delay. Problems with her bowel movements had been recognized since she was about 3-year-old. Analysis of her rectal biopsy revealed no pathological findings indicative of Hirschsprung disease. At the same time, Ehlers-Danlos syndrome was also suspected (ED score: 4 points), but electron microscopy analysis of her skin biopsy showed no obvious findings. She was hence treated with laxatives, such as oxidized Mg and laxoberon, but she only had bowel movements once every 3 days to 5 days. At 13 years of age, she was admitted again for disimpaction. Her height was 134 cm (--3.8 SD), and her weight was 24 kg (--2.9 SD). Her fingers were slender, and webbing was observed between the fingers. She had hyperextension of the joints and facial anomalies (constant open mouth, thin and prominent jaw and nose, and raised ears). An enema, disimpaction, and an abdominal CT scan were performed. The CT displayed extreme megacolon that had displaced the bladder (Figure 2). Brain CT and MRI displayed no abnormalities. Her constipation improved after the treatment. After discharge, her condition was well controlled by laxatives. Her elder and younger brothers both had mental retardation IQ of the younger brother was 43 by WSIC-III. Both brothers had shown a tendency of constipation from 1-year-old. The brothers also visited our department because of their severe constipation and abdominal fullness, and they were hospitalized for examination at 12 years of age. The elder brother underwent a rectal mucosal biopsy, but Acetyl Cholinesterase (AChE) staining revealed that there was no proliferation of nerve fibers. Treatment with laxatives, such as oxidized Mg and laxoberon® was started, and they have since then experienced daily bowel movements. The younger brother also developed obesity and liver dysfunction at the age of 11 years, in addition to constipation. His liver biopsy showed chronic hepatitis and fatty liver, and he was diagnosed as having nonalcoholic steatohepatitis. Histologically, the rectal mucosa and superficial submucosal tissue showed mild chronic inflammatory cell infiltration within the mucosa. There was no proliferation of nerve fibers, which were positive for AChE staining. The biopsy samples showed no characteristics indicative of Hirschsprung disease. Brain MRI displayed mild atrophy without any vascular anomalies. All characteristics were shown in (Table 1).

Methods
Genomic Studies
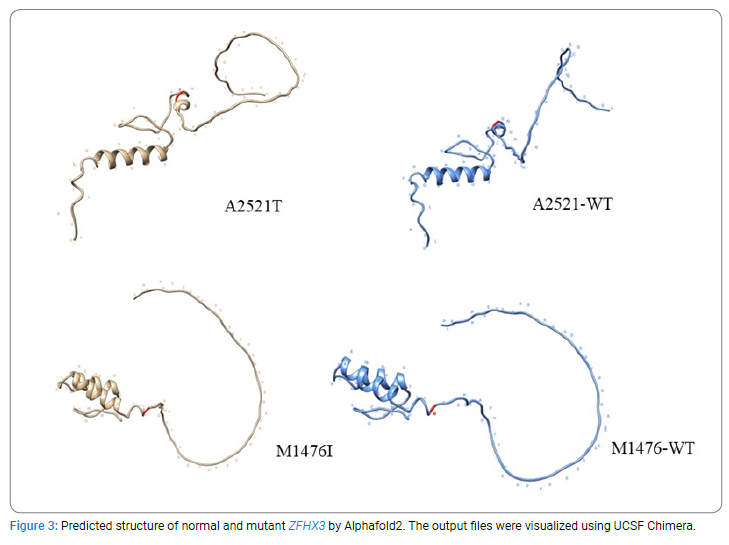
Exome sequencing on blood samples from the 3 siblings was performed using Ion Proton sequencer. The effects of the detected variations on protein structure and function were mostly analyzed by SIFT and PolyPhen2. Alphafold2 was also used to predict the structure of the normal type and mutant type [6]. The output files were visualized using UCSF Chimera [7]. To understand how to evaluate the effects of mutations by AI, Alphamissense software was also used [8]. According to the results of exome sequencing and the damaging effects of the vairants, ZFHX3 A2530T (which corresponds to human A2521T) knockout mice were created, and their neurological behavior was analyzed. C57BL/6N mice were purchased from CLEA Japan. The mutant mice generated by the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats / CRISPR associated proteins) system were maintained on the C57/BL6N background at Riken BioResource Research Center (BRC). We applied a standard procedure of the CRISPR/Cas9 system to introduce the pathogenic ZFHX3 variant into the mouse genome.
The crRNA was designed to target the sequence 5′-CACGTCGACCCCTCAACAGCTGG-3′ in the mouse ZFHX3 locus. The gRNA was prepared by mixing equimolar amounts of crRNA and tracrRNA (Integrated DNA Technologies, Iowa 52241, USA). The gRNA duplex was mixed with 200 ng/μL of the Cas9 protein (Integrated DNA Technologies) and 200 ng/μL of a single stranded oligonucleotide donor in HEPES-buffered saline, and this was then introduced into fertilized eggs using CUY21EDIT II electroporator (Bex, Japan). After electroporation, eggs were cultured in KSOM medium (Arc Resource, Japan) for 1 day, and then transferred into the oviducts of recipient female mice. The sequence of the 120-nt single stranded oligonucleotide donor was 5΄-CCTCGCAGCTCTCCCATCTGCCCCTCAAGCCCCTCCACACGTCG ACCCCTCAACAGCTGaCAAATACCTCCTCAGCTAATCCCCTACCAGTGTGCAAGCTGGCGTGGGGTTCCCAT-3΄. Phenotype data of mutant mice were collected and analyzed by the comprehensive phenotyping platform of Japan Mouse Clinic at Riken BRC (https://ja.brc.riken.jp/lab/jmc/mouse_clinic/en/index.html). All animal experiments in this study were performed in accordance with the guidelines approved by the Animal Care and Use Committee of Riken BRC.
Results
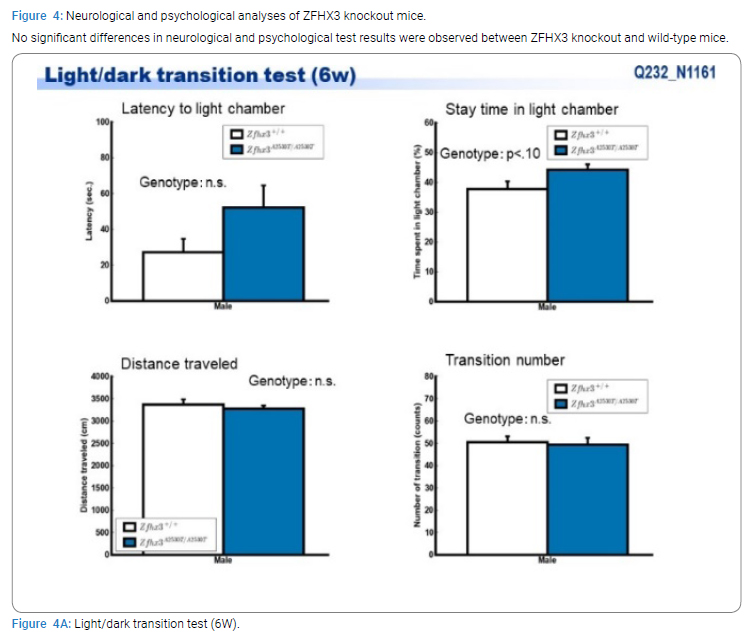
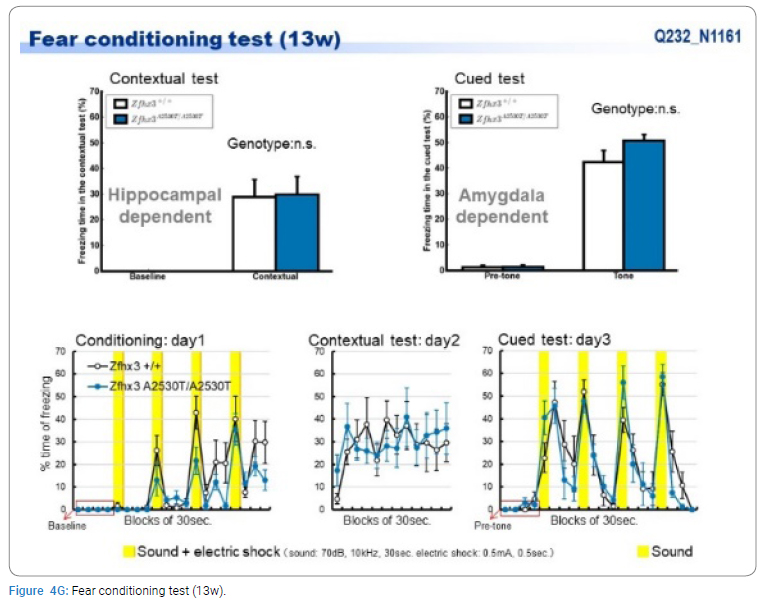
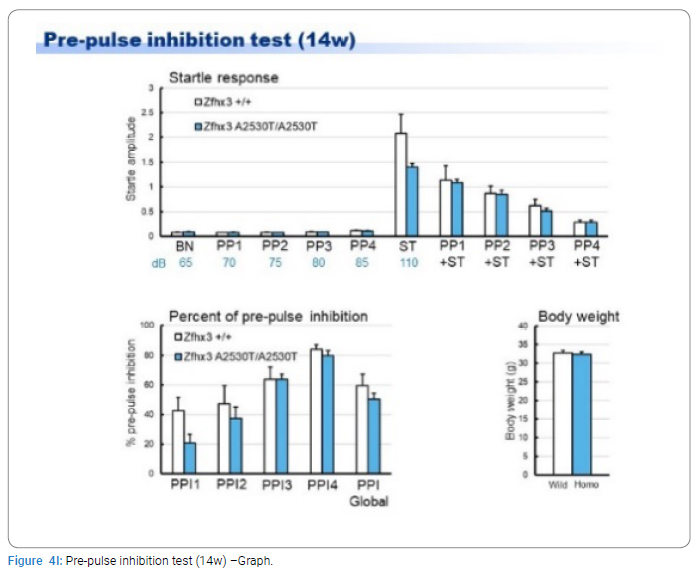
Exome sequencing of the 3 siblings identified 2 variants of ZFHX3 (c.G756A:p.A2521T and c.G4428A:p.M1476I) as candidates responsible for their diseases, as a compound-heterozygous model, as shown in (Table 2). The former mutation (A2521T) was shown to be damaging by SIFT and PolyPhen2. We analyzed the results of 100 residues, including the variants, to clarify the 3D structure of the variant proteins (Figure 3). However, it was difficult to evaluate the effects of these variants by Alphafold2, which predicts the structure of the normal and mutant. Alphamissense (AI) rated A2521T and M1476I as benign. The resultant mice with a frameshift in ZFHX3 were fetal lethal. Mice with knockout did not show lethality, and their neurological and psychological characteristics were analyzed. These mice showed no significant difference with wild-type mice in all of the various neurological tests (light/dark transition test, open-field test, Crawley social interaction test, home-cage activity test, Y-maze, fear conditioning test, and pre-pulse inhibition test), as shown in (Figure 4A–4I).


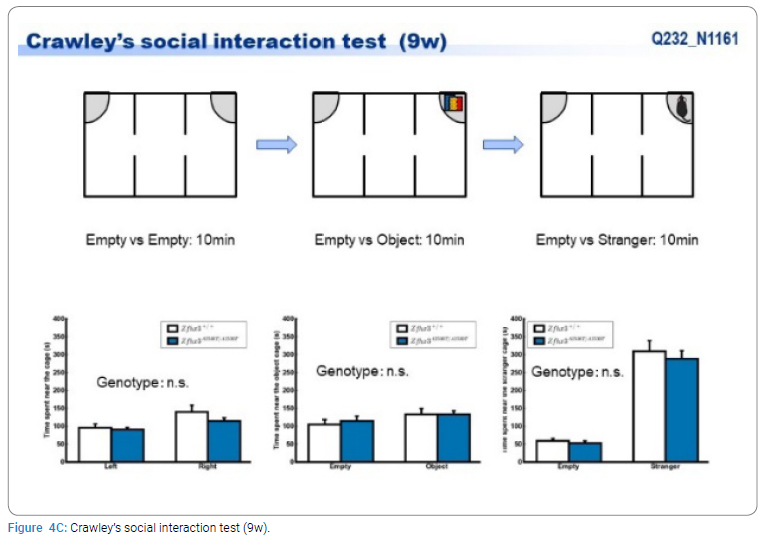
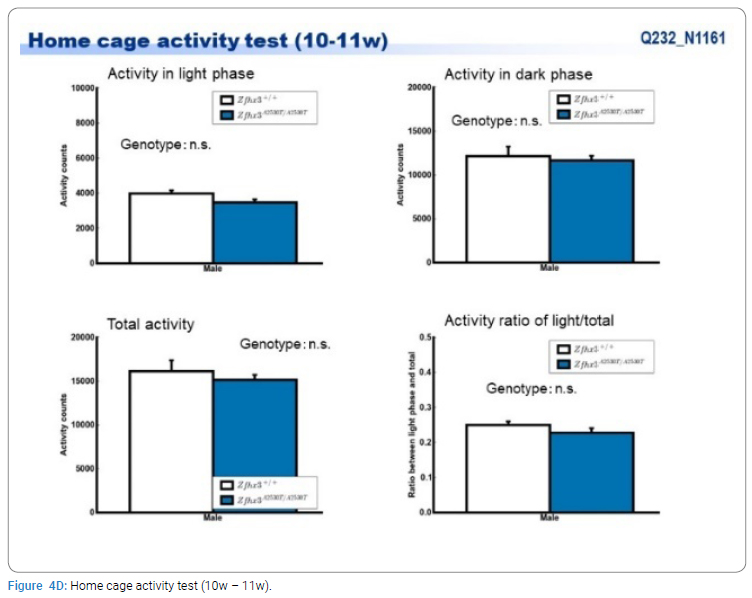
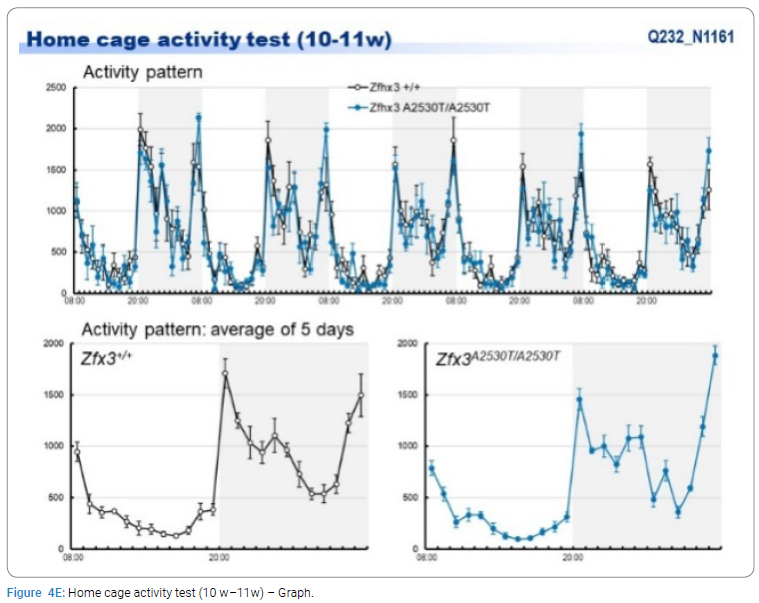
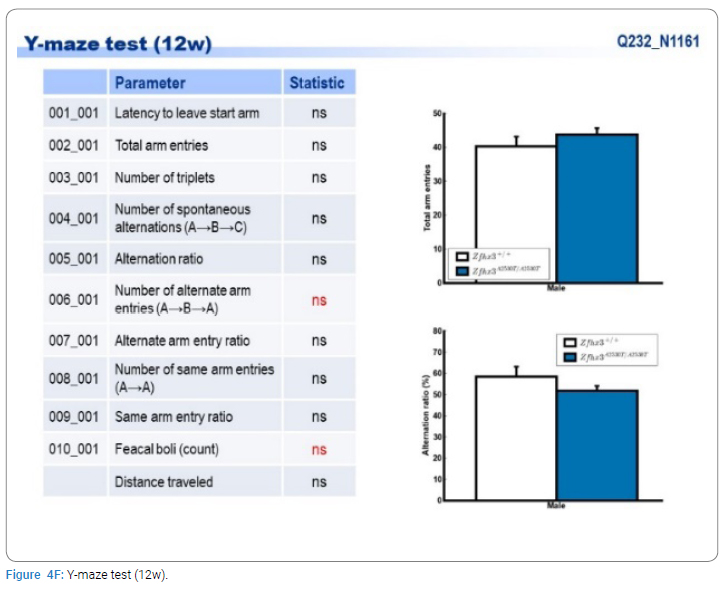

Discussion
AAS, which is a rare syndrome with a recessive mode of inheritance, was first described by Aarskog in 1970, and then further described in detail by Scott in 2 different families. The characteristics of AAS individuals were reported as short-statured with craniofacial anomalies (hypertelorism, short nose, ptosis, and genital dysmorphism, such as shawl scrotum and cryptorchidism) [9,10]. Various other characteristics, such as clinodactyly, brachydactyly, long philtrum, widow’s peak, camptodactyly, interdigital webbing, and inguinal/umbilical hernia have also been reported [11]. Fifty-two pathogenic variants of FGD1, which is the responsible gene for the X-linked form of AAS, have been reported [12]. However, no clear phenotype-genotype correlation has been found with mutations in FGD [13]. Although AAS is clinically and genetically heterogeneous, causative genes other than FGD1 have not yet been identified. In this study, we identified ZFHX3 as a new candidate causative gene of AAS. Zhang et al. performed a whole-exome sequencing study and isolated a panel of genes as candidate causative genes of Hirschsprung’s disease, and reported that ZFHX3 was prioritized for follow-up studies: both the time-space expression patterns in the mouse and human colon showed that it is a strong candidate gene for Hirschsprung’s disease [14]. Zhang et al. demonstrated that the striatum, which is the major component of the basal ganglia, consists of the caudate-putamen, nucleus accumbens, and olfactory tubercle. The striatal principal projection neurons are comprised of Medium Spiny Neurons (MSNs) with 2 dopamine receptors, namely DRD1 (Dopamine receptor D1) (D1 MSNs) and DRD2 (D2 MSNs). ZFHX3 is strongly expressed in the boundary of the sub ventricular zone / mantle zone of the lateral ganglionic eminence and its expression in the striatum is down regulated during the first postnatal week. At the cellular level, ZFHX3 is selectively expressed in immature D1 MSNs. Moreover, a significant reduction in the number of D1 MSNs in the striatum of ZFHX3 conditional knockout mice was observed. They concluded that ZFHX3 plays a crucial role in the differentiation and survival of late-born D1 MSNs [15]. Regarding the neurological and psychological manifestations of individuals with AAS, individuals have been reported to show an average IQ in most cases. However, various degrees of neurocognitive disabilities and/or behavior disorders, ranging from attention-deficit/hyperactivity disorder to severe intellectual disability were reported [16–19]. ZFHX3 is also known as ATBT/ATBF1, and has been reported in several studies of the nervous system especially neuronal differentiation [20,21]. Further genomic and pathological studies should be performed in the future. We could not find any study reporting neurological manifestations in ZFHX3 knockout mice, nor a pathology similar to Hirsch sprung disease in AAS patients. From these reports, ZFHX3 is assumed to be associated with neuronal differentiation. In the present study, all affected individuals had mental retardation. Other effects caused by having a megacolon for a long time, including effects on the nutritional environment, might occur as the individuals grow and develop. In the present study, mice with a frame shift in ZFHX3 were fetal lethal, whereas mice with A2530T (which corresponds to human A2521T) knockout did not show lethality. We speculated that the frame shift mutation may have a more severe effect on the protein’s function than the A2530T mutation. However, further research is needed to confirm this hypothesis. To our knowledge, there are no direct lines of evidence to date that ZFHX3 can cause specific diseases. Somatic mutations in ZFHX3 have been detected in prostate cancers. The most frequently mutated genes in prostate cancer are KMT2D (26.45%), FOXA1 (16.13%), ATM (15.81%), ZFHX3 (9.35%), TP53 (8.06%), and APC (5.48%). Hotspot mutations in ZFHX3 have been identified in human prostate cancer [22]. Variants of ZFHX3 have also been reported to be associated with atrial fibrillation, cerebral infarction, and lung thromboembolism. A recent study identified a loss-of-function variation in ZFHX3 as a novel cause of syndromic Intellectual Disability (ID). They found that the loss-of-function variation of ZFHX3 consistently associates with (mild) ID and/or behavioral problems, postnatal growth retardation, feeding difficulties, and recognizable facial characteristics, including the rare occurrence of cleft palate [23]. Our present patients with variants of ZFHX3 are similar to their cases. Unfortunately, we did not investigate pathological changes occurring in the central nervous system of A2530T knockout mice. Further studies on these effects are needed to make any conclusions regarding the association between ZFHX3 and neuronal differentiation.
Conclusion
We found a new candidate disease-causing gene in 3 siblings who have Aarskog-Scott syndrome with mental retardation and megacolon. Mice with a frame shift mutation in ZFHX3 were fetal lethal. However, mice with a knockout mutation in ZFHX3 were viable and did not have any obvious neurological abnormalities. An association between the ZFHX3 mutation and their symptoms was not clear.
Authors Contributions
H.K. and N.S. designed the study; SS. and MY. Performed the exome analysis, TN. and YK. Collected the data, and HK. Wrote the manuscript; AT., TM., AA and NK. Performed the knockout mouse experiments, including the behavioral analysis. All authors read and approved the final manuscript. H.K. critically reviewed the manuscript and supervised the whole study process. All authors read and approved the final manuscript.
Acknowledgment
Not applicable.
Funding
This research received no specific grants from any funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Porteous ME, Goudie DR. Aarskog syndrome. J Med Genet. 1991;28(1):44–47.
- Orrico A, Galli L, Faivre L, Clayton-Smith J, Azzarello-Burri SM, Hertz JM, et al. Aarskog-Scott syndrome: clinical update and report of nine novel mutations of the FGD1 gene. Am J Med Genet A. 2010;152A(2):313–318.
- Orrico A, Galli L, Cavaliere ML, Garavelli L, Fryns JP, et al. Phenotypic and molecular characterisation of the Aarskog-Scott syndrome: a survey of the clinical variability in light of FGD1 mutation analysis in 46 patients. Eur J Hum Genet. 2004;12(1):16–23.
- Fryns JP. Dolichomegasigmoid in Aarskog syndrome. Am J Med Genet. 1993;45(1):122.
- Casteels M, Samain H, Penninckx F, Coremans G, Beirinckx J, Fryns JP. Megadolichosigmoid in a young male with Aarskog syndrome. Genet Couns. 1994;5(1):81–83.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
- Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023;381(6664):eadg7492.
- Aarskog D. A familial syndrome of short stature associated with facial dysplasia and genital anomalies. J Pediatr. 1970;77(5):856–861.
- Scott CI. Unusual facies, joint hypermobility, genital anomaly and short stature: a new dysmorphic syndrome. Birth Defects Orig Artic Ser. 1971;7(6):240–246.
- Schwartz CE, Gillessen-Kaesbach G, May M, Cappa M, Gorski J, Steindl K, et al. Two novel mutations confirm FGD1 is responsible for the Aarskog syndrome. Eur J Hum Genet. 2000;8(11):869–874.
- Zanetti Drumond, Sousa Salgado L, Sousa Salgado C, Oliveira VAL, de Assis EM, Campos Ribeiro M, et al. The prevalence of clinical features in patients with aarskog-scott syndrome and assessment of genotype-phenotype correlation: a systematic review. Genet Res (Camb). 2021:2021:6652957.
- Pérez-Coria M, Lugo-Trampe JJ, Zamudio-Osuna M, Rodríguez-Sánchez IP, Lugo-Trampe A, de la Fuente-Cortez B, et al. Identification of novel mutations in Mexican patients with Aarskog-Scott syndrome. Molecular Genetics & Genomic Medicine, 2015;3(3):197–202.
- Zhang Z, Li Q, Diao M, Liu N, Cheng W, Xiao P, et al. Sporadic hirschsprung disease: mutational spectrum and novel candidate genes revealed by next-generation sequencing. Sci Rep. 2017;7(1):14796.
- Zhang Z, Wei S, Du H, Su Z, Wen Y, Shang Z, et al. Zfhx3 is required for the differentiation of late born D1-type medium spiny neurons. Exp Neurol. 2019;322:113055.
- Kaname T, Yanagi K, Okamoto N, Naritomi K. Neurobehavioral disorders in patients with Aarskog-Scott syndrome affected by novel FGD1 mutations. Amer Jour of Med Gen Part A. 2006;140(12):1331–1332.
- Lebel RR, May M, Pouls S, Lubs HA, Stevenson RE, Schwartz CE. Non-syndromic X-linked mental retardation associated with A missense mutation (P312L) in the FGD1 gene. Clinical Genetics. 2002;61(2):139–145.
- Logie LJ, Porteous ME. Intelligence and development in Aarskog syndrome. Archives of Disease In Childhood. 1998;79(4):359–360.
- Orrico A, Galli L, Buoni S, Hayek G, Luchetti A, Lorenzini S, et al. Attention-deficit/hyperactivity disorder (ADHD) and variable clinical expression of Aarskog-Scott syndrome due to A Novel FGD1 gene mutation (R408Q). AmcaJurnl of Med Gen Part A. 2005;135(1)99–102.
- Ido A, Miura Y, Tamaoki T. Activation of ATBF1, a multiple-homeodomain zinc-finger gene, during neuronal differentiation of murine embryonal carcinoma cells. Dev Biol. 1994;163(1):184–187.
- Moreau MX, Saillour Y, Cwetsch AW, Pierani A, Causeret F. Single-cell transcriptomics of the early developing mouse cerebral cortex disentangle the spatial and temporal components of neuronal fate acquisition. Dev. 2021;148(14):dev197962.
- Mangolini A, Rocca C, Bassi C, Ippolito C, Negrini M, Dell'Atti L, et al. Detection of disease-causing mutations in prostate cancer by NGS sequencing. Cell Biol Int. 2022;46(7):1047–1061.
- Del Rocío Pérez Baca M, Jacobs EZ, Vantomme L, Leblanc P, Bogaert E, Dheedene A, et al. A novel neurodevelopmental syndrome caused by loss-of-function of the Zinc Finger Homeobox 3 (ZFHX3) gene. medRxiv. 2023;2023.05.22.23289895.
Keywords
ZFHX3; Neuron; Constipation; Next generation sequencing
Cite this article
Kawashima H, Nishimata S, Shinji S, Morishima Y, Tsutsumi N, Kashiwagi K, et al. Three siblings with aarskog scott syndrome together with mental retardation and giant megacolon. Glob J Pedia. 2024;2(1):1–11.
